
Bekanntmachung einer Mitteilung zum Homöopathischen Arzneibuch (Empfehlungen der Fachausschüsse der Deutschen Homöopathischen Arzneibuch-Kommission)
Bundesinstitut
für Arzneimittel und Medizinprodukte
Bekanntmachung
einer Mitteilung
zum Homöopathischen Arzneibuch
(Empfehlungen der Fachausschüsse der
Deutschen Homöopathischen Arzneibuch-Kommission)
Auf Grund des § 7 Absatz 5 der Geschäftsordnung für die Deutsche Homöopathische Arzneibuch-Kommission und deren Gremien (Bekanntmachung vom 17. Juli 2009, BfArM-Internetseite) sind Empfehlungen der Fachausschüsse der Deutschen Homöopathischen Arzneibuch-Kommission den betroffenen Fach- und Wirtschaftskreisen zur Kenntnis zu bringen.
Der Fachausschuss Analytik der Deutschen Homöopathischen Arzneibuch-Kommission hat die unten genannten Entwürfe für neue und revidierte Monographien für die Aufnahme in das Homöopathische Arzneibuch empfohlen. Sie werden hiermit zur Kenntnis gebracht (Anlage).
Neue Monographien
- 1.
-
Aspidosperma quebracho-blanco (Quebracho)
- 2.
-
Ferrum picrinicum
- 3.
-
Juglans regia (Juglans)
Revidierte Monographien
- 1.
-
Allium sativum
- 2.
-
Arsenum iodatum (Arsenum jodatum)
- 3.
-
Blatta orientalis
- 4.
-
Chelidonium majus (Chelidonium)
- 5.
-
Chelidonium majus e floribus, ethanol. Digestio (Chelidonium, Flos, ethanol. Digestio)
- 6.
-
Chelidonium majus Rh (Chelidonium Rh)
- 7.
-
Hydrargyrum stibiato-sulfuratum (Aethiops antimonialis)
- 8.
-
Stibium arsenicosum (Antimonium arsenicosum)
- 9.
-
Stibium sulfuratum aurantiacum (Antimonium sulfuratum aurantiacum)
- 10.
-
Stibium sulfuratum nigrum
Stellungnahmen zu dem oben genannten Entwurf des Homöopathischen Arzneibuches sind bis spätestens 21. September 2023 einschließlich an die Geschäftsstelle der Arzneibuch-Kommissionen im Bundesinstitut für Arzneimittel und Medizinprodukte, Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn, zu richten.
43.07-2023-24178
Bundesinstitut
für Arzneimittel und Medizinprodukte
In Vertretung
Knöß
Neue Monographien
Aspidosperma quebracho-blanco
Quebracho
Definition
Verwendet wird die getrocknete Stamm- und Zweigrinde von Aspidosperma quebracho-blanco Schltdl, die bezogen auf die getrocknete Droge mindestens 0,70 Prozent (m/m) Alkaloide, berechnet als Yohimbin (C21H26N2O3; Mr 354,4) enthält.
Beschreibung
Die Rindenstücke sind 2 bis 3 cm dick, hart, flach oder rinnenförmig, außen tief gefurcht und querrissig. Die Innenrinde ist gelblich grau, rötlich grau oder gelbbraun und deutlich längsstreifig, der Bruch ist kurzsplitterig. Der Querschnitt zeigt eine mächtige, gelblich- bis ziegelrote, von zahlreichen hellen Bändern durchzogene Borke, die sich scharf von der Innenrinde abhebt. Borke und Rinde erscheinen gelblich weiß gesprenkelt.
Schnittdroge: Die Schnittdroge ist gekennzeichnet durch gelblich- bis ziegelrote Borkenstückchen und rötlich graue bis gelbbraune Rindenteile. Die Droge erscheint weißlich punktiert.
Mikroskopische Merkmale: Es ist kein primäres Rindengewebe vorhanden. Die Borke der sekundären Rinde ist von zahlreichen Korkfragmenten durchzogen, die aus dünnwandigen, farblosen, unverholzten Zellen bestehen. Zwischen den Korkfragmenten und in den inneren Rindenschichten befinden sich zahlreiche Bündel dickwandiger Sklerenchymfasern und in Nestern vorliegende dickwandige, getüpfelte Steinzellen. Sowohl Sklerenchymfasern als auch Steinzellen sind umhüllt von Kristallzellreihen von Calciumoxalatkristallen. Zusätzlich sind auch zahlreiche frei liegende Kristalle vorhanden. Die sekundäre Rinde ist von zwei- bis dreireihigen Markstrahlen durchzogen. Im dünnwandigen Parenchym können sich einfach oder zwei- bis vierfach zusammengesetzte Stärkekörner befinden. Die inneren Rindenschichten weisen meist kollabierte Siebröhren mit leitersprossenartig versteiften Siebplatten auf.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: 1,0 g pulverisierte Droge (355) wird mit 10 ml Salzsäure 1 % RN versetzt und für 10 Minuten im Wasserbad erhitzt. Anschließend wird die Mischung mit 0,5 g wasserfreiem Natriumcarbonat R versetzt und nach Beendigung der Gasentwicklung filtriert. Das Filtrat wird dreimal mit je 25 ml Dichlormethan R extrahiert. Die vereinigten organischen Phasen werden über wasserfreiem Natriumsulfat R getrocknet, unter vermindertem Druck zur Trockne eingeengt und der Rückstand in 1 ml Methanol R aufgenommen.
Referenzlösung: 5 mg Yohimbinhydrochlorid RN und 5 mg Noscapinhydrochlorid R werden jeweils in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R (5 bis 40 μm) [oder DC-Platte mit Kieselgel R (2 bis 10 μm)]
Fließmittel: Diethylamin R, Ethylacetat R, Toluol R (10:20:70 V/V/V)
Auftragen: 50 μl [oder 10 μl] Untersuchungslösung und 20 μl [oder 5 μl] Referenzlösung; bandförmig 20 mm [oder 8 mm].
Laufstrecke: 10 cm [oder 6 cm]
Trocknen: 100 bis 105 °C bis zum Verschwinden des Fließmittelgeruchs
Detektion: Die Platte wird mit verdünntem Dragendorffs Reagenz R und anschließend mit Natriumnitrit-Lösung R besprüht. Die Auswertung erfolgt im Tageslicht.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
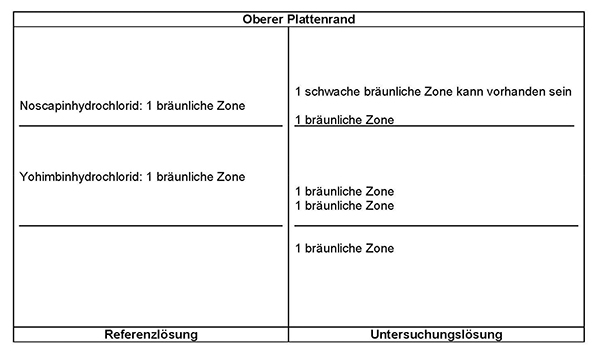
Prüfung auf Reinheit
Fremde Bestandteile (2.8.2): höchstens 2 Prozent
Trocknungsverlust (2.2.32): höchstens 12,0 Prozent, mit 1,000 g pulverisierter Droge (355) durch 2 h langes Trocknen im Trockenschrank bei 100 bis 105 °C bestimmt
Asche (2.4.16): höchstens 10,0 Prozent
Gehaltsbestimmung
1,000 g pulverisierte Droge (335) werden mit 50 ml Salzsäure 1 % RN 30 min lang im Wasserbad erhitzt. Nach dem Erkalten wird die Mischung mit Wasser R zu 100,0 ml ergänzt und durch Watte filtriert. Die ersten 25 ml Filtrat werden verworfen. Von dem weiteren Filtrat werden 15,0 ml mit 0,5 g wasserfreiem Natriumcarbonat R versetzt und nach Beendigung der Gasentwicklung auf ein mit 15 g granulierter Kieselgur RH gefülltes Chromatographierohr von etwa 150 mm Länge und 30 mm innerem Durchmesser gegeben1 und der Kolben mit 3 ml Wasser R nachgewaschen. Die Waschflüssigkeit wird ebenfalls auf die Säule gegeben und nach 15 min wird mit 80 ml Dichlormethan R eluiert. Nach dem Abtropfen des Dichlormethans wird das Eluat unter vermindertem Druck auf etwa 20 bis 30 ml eingeengt und mit Dichlormethan R auf 50,0 ml ergänzt. 30,0 ml dieser Lösung werden in einen Scheidetrichter pipettiert und mit 20,0 ml Citrat-Pufferlösung pH 4,0 RN und 5,0 ml Eriochromschwarz-T-Lösung RN versetzt. Die Mischung wird kräftig geschüttelt. Eventuell auftretende Emulsionen werden durch Zugabe von 1 ml gesättigter Natriumchlorid-Lösung R beseitigt. Die rot gefärbte, organische Phase wird durch einen mit Dichlormethan befeuchteten Filter in einen 100-ml-Messkolben filtriert, der 10,0 ml Methanol R enthält. Die wässrige Phase wird noch 3-mal mit je 20 ml Dichlormethan R extrahiert. Die vereinigten organischen Phasen werden mit Dichlormethan R zu 100,0 ml verdünnt.
Die Absorption A (2.2.25) der Lösung wird bei 520 nm gegen Dichlormethan R gemessen.
Der Prozentgehalt (m/m) an Alkaloiden, berechnet als Yohimbin, wird mit Hilfe der spezifischen Absorption 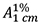 = 495 nach folgender Formel berechnet:
= 495 nach folgender Formel berechnet:
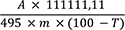
m = Einwaage der Droge in Gramm
T = Trocknungsverlust in Prozent
Arzneiformen
Die Urtinktur enthält mindestens 0,020 und höchstens 0,090 Prozent (m/m) Alkaloide, berechnet als Yohimbin (C21H26N2O3; Mr 354,5).
Herstellung
Urtinktur aus der pulverisierten Droge (710) und flüssige Verdünnungen nach Vorschrift 4a mit Ethanol 62 % (m/m)
Eigenschaften
Die Urtinktur ist eine rotbraune Flüssigkeit.
Prüfung auf Identität
Die Urtinktur gibt die Identitätsreaktion der Droge.
Untersuchungslösung: 10 ml Urtinktur werden unter vermindertem Druck zur Trockne eingedampft. Der Rückstand wird in 1 ml Methanol R aufgenommen und filtriert.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
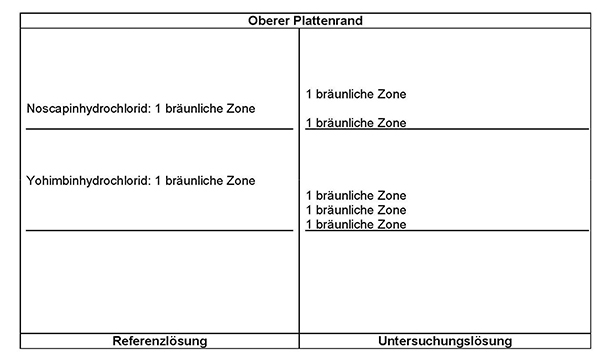
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,880 bis 0,900
Trockenrückstand (H 2.2.6): mindestens 0,3 Prozent
Gehaltsbestimmung
2,000 g Urtinktur werden unter vermindertem Druck zur Trockne eingeengt. Der Rückstand wird mit 5 ml Wasser R versetzt und nach Zugabe von 0,5 g wasserfreiem Natriumcarbonat R bis zum Lösen im Ultraschallbad behandelt. Die Mischung wird in ein mit 15 g granulierter Kieselgur RH gefülltes Chromatographierohr von etwa 150 mm Länge und 30 mm innerem Durchmesser gegeben2 und der Kolben dreimal mit je 4 ml Wasser R nachgewaschen. Die Waschflüssigkeit wird ebenfalls auf die Säule gegeben und nach 15 min wird mit 80 ml Dichlormethan R eluiert.
Die weitere Bestimmung erfolgt wie bei der Droge angegeben.
Der Prozentgehalt (m/m) an Alkaloiden, berechnet als Yohimbin, wird mit Hilfe der spezifischen Absorption = 495 nach folgender Formel berechnet:
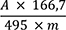
m = Einwaage der Urtinktur in Gramm
Ferrum picrinicum
| C12H4N6O14Fe ∙ 5 H2O | M r 602,0 |
Verwendet wird die Substanz, die durch die Reaktion von Acidum picrinicum für homöopathische Zubereitungen (Ph. Eur.), Calciumcarbonat (Ph. Eur.) und Eisen(II)-sulfat-Heptahydrat (Ph. Eur.) erhalten wurde. Sie enthält mindestens 0,10 und höchstens 0,12 Prozent Eisen (Mr 55,85).
Arzneiformen
Die Substanz entspricht der Lösung D2.
Die 3. Dezimalverdünnung wird aus 1 Teil der Lösung D2 und 9 Teilen Ethanol 15 % (m/m), die folgenden Verdünnungen nach Vorschrift 5a mit Ethanol 43 % (m/m) hergestellt.
Herstellung
4,58 Teile Acidum picrinicum für homöopathische Zubereitungen, berechnet auf die getrocknete Substanz, werden in 300 Teilen Gereinigtem Wasser (Ph. Eur.) unter Erhitzen vollständig gelöst. Nach dem Abkühlen wird unter starkem Schütteln zu dieser Lösung 1,0 Teil Calciumcarbonat (Ph. Eur.) hinzugefügt. Die Mischung wird solange erhitzt und geschüttelt, bis sich kein Kohlendioxid mehr bildet. Nach dem Abkühlen auf Raumtemperatur werden dem Ansatz eine Lösung aus 2,80 Teile Eisen(II)-sulfat-Heptahydrat (Ph. Eur.), gelöst in 50 Teilen Gereinigtem Wasser (Ph. Eur.), zugesetzt. Der Ansatz wird gut durchmischt, und mit 81,0 Teilen Ethanol 96 % (Ph. Eur.) versetzt und wiederum gut durchmischt.
Nach Lagerung des Ansatzes für etwa 48 bis 72 Stunden im Kühlschrank (2–8 °C) wird das ausgefallene Calciumsulfat mit Hilfe eines Filters für langsame Filtration abfiltriert. Das erhaltene Filtrat wird gewogen und mit Gereinigtem Wasser (Ph. Eur.) auf 512,0 Teile aufgefüllt und gut durchmischt.
Eigenschaften
Klare, intensiv gelb gefärbte Flüssigkeit; in jedem Verhältnis mischbar mit Ethanol und Wasser
Prüfung auf Identität
- A.
-
Die Substanz gibt die Identitätsreaktion a auf Eisen (2.3.1).
- B.
-
Wird 1 ml der Substanz mit 0,2 ml verdünnter Natriumhydroxid-Lösung R versetzt, entsteht ein brauner Niederschlag.
- C.
-
Wird 1 ml der Substanz mit 5 ml Wasser R und 0,5 ml Kaliumcyanid-Lösung R versetzt, färbt sich die Lösung intensiv rot.
- D.
-
1 ml Substanz wird in einem Becherglas mit 30 mg Kaliumnitrat R und 1 ml konzentrierter Natriumhydroxid-Lösung R versetzt. Die Mischung wird zum Sieden über einem Bunsenbrenner erhitzt. Wird anschließend sofort über die Öffnung des Becherglases ein rotes, mit Wasser R befeuchtetes Lackmuspapier gehalten, färbt sich das Lackmuspapier blau.
Prüfung auf Reinheit
Aussehen der Lösung: Die Substanz muss klar sein (2.2.1).
Sulfat (2.4.13): 0,30 g der Substanz werden mit destilliertem Wasser R zu 15 ml verdünnt.
Die Lösung muss der Grenzprüfung auf Sulfat entsprechen (500 ppm).
Relative Dichte (2.2.5): 0,981–0,986
Gehaltsbestimmung
20,00 g der Substanz werden mit einer Mischung von 300 ml Wasser R und 20 ml konzentrierter Schwefelsäure R versetzt. Nach Zusatz von 0,5 ml Ferroin-Lösung R wird die Lösung mit Cer(IV)-sulfat-Lösung (0,1 mol ∙ l–1) bis zum Farbumschlag von rot nach hellgrün titriert.
1 ml Cerium(IV)-sulfat-Lösung (0,1 mol ∙ l–1) entspricht 5,585 mg Eisen
Lagerung
Vor Licht geschützt und kühl lagern (unter 15 °C).
Juglans regia
Juglans
Verwendet werden zu gleichen Teilen die frischen, grünen Fruchtschalen und die Blätter von Juglans regia L.
Beschreibung
Die Laubblätter sind unpaarig gefiedert und in der Jugend rötlich. Der Blattstiel ist bis zu 35 cm lang und am Grunde stark angeschwollen. Das Blatt besteht aus 5 bis 9 (meist 7), 5 bis 13 (maximal 19) cm langen und bis 5 (maximal 8) cm breiten, breitelliptischen und ganzrandigen Fiedern. Sie sind glänzend grün, in der Jugend drüsig punktiert und in den Aderachseln gebärtet. Die lang gestielten Endfiedern sind etwas größer und bis zu 25 cm lang. Die Fruchtschale ist außen glatt, grün, mit weißlicher Punktierung und löst sich in unregelmäßigen, zäh fleischigen Stücken ab.
Arzneiformen
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 3a
Eigenschaften
Die Urtinktur ist eine dunkelbraune Flüssigkeit.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: die Urtinktur
Referenzlösung: 10 mg Hyperosid R und 5 mg Kaffeesäure R werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R (5 bis 40 μm) [oder DC-Platte mit Kieselgel R (2 bis 10 μm)]
Fließmittel: Ameisensäure R, Wasser R, Ethylacetat R (6:6:88 V/V/V)
Auftragen: 20 μl [oder 5 μl] Untersuchungslösung und 10 μl [oder 3 μl] Referenzlösung; bandförmig 20 mm [oder 10 mm]
Laufstrecke: 15 cm [oder 6 cm]
Trocknen: 5 min lang in einem Luftstrom bei Raumtemperatur
Detektion: Die Platte wird 5 min lang bei 100 bis 105 °C erhitzt; die warme Platte wird mit einer Lösung von DiphenylboryloxyethylaminR (10 g · l–1) in Methanol R und anschließend mit einer Lösung von Macrogol 400 R (50 g · l–1) in Methanol R behandelt. Die Platte wird etwa 30 min lang an der Luft trocknen gelassen. Anschließend erfolgt die Auswertung im ultravioletten Licht bei 365 nm.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
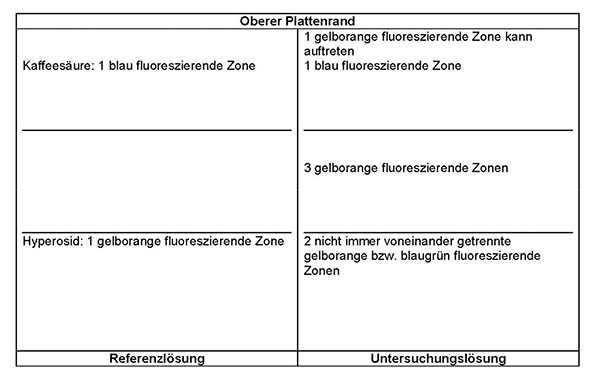
Prüfung auf Reinheit:
Relative Dichte (2.2.5): 0,900 bis 0,925
Trockenrückstand (H 2.2.6): mindestens 2,0 Prozent
Gesamt-Chinon-Derivate (2.2.25): höchstens 0,017 Prozent Gesamt-Chinon-Derivate, berechnet als Juglon (C10H6O3)
Untersuchungslösung:
5,000 g Urtinktur werden mit 10 ml verdünnter Salzsäure R und 2,5 ml Eisen(III)-chlorid-Lösung R1 versetzt und 30 Minuten lang im Wasserbad unter Rückfluss erhitzt. Die Lösung wird abgekühlt, in einen Scheidetrichter überführt und zweimal mit 20 ml Pentan R ausgeschüttelt. Die vereinigten organischen Lösungen werden durch ein Filter3 filtriert. Das Filtrat wird unter vermindertem Druck zur Trockne eingeengt und der Rückstand in 20,0 ml Ethanol 96 % R aufgenommen.
Die Absorption (2.2.25) dieser Lösung wird gegen Ethanol 96 % R als Kompensationsflüssigkeit bei 422 nm gemessen.
Der Prozentgehalt (m/m) an Gesamt-Chinon-Derivaten, berechnet als Juglon, wird mit Hilfe der spezifischen Absorption = 214 nach folgender Formel berechnet:
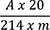
A = Absorption der Untersuchungslösung
m = Einwaage der Urtinktur in Gramm
Lagerung
Vor Licht geschützt.
Revidierte Monographien
Anmerkungen zur Monographie:
- –
-
Herstellung: Die Mazerationszeit wird auf die Standard-Mazerationszeit von 10 bis 30 Tagen geändert, da in der Herstellungspraxis eine exakte Einhaltung von 10 Tagen nicht immer möglich ist und belegt wurde, dass sowohl innerhalb der Standard-Mazerationszeit als auch bei der längeren und sehr langen Mazerationszeit keine signifikanten Unterschiede bei den Ergebnissen der analytischen Prüfungen, wie im DC und bei Trockenrückstand und relativer Dichte, auftraten.
- –
-
Prüfung auf Reinheit: Die relative Dichte wird basierend auf einer repräsentativen Anzahl von Chargendaten verschiedener Hersteller auf 0,890 bis 0,935 geändert.
Allium sativum
Die frischen Zwiebeln müssen der Monographie Allium sativum für homöopathische Zubereitungen (Ph. Eur.) entsprechen.
Arzneiformen
Die Urtinktur muss bis auf die Herstellung und die relative Dichte der Monographie Allium sativum für homöopathische Zubereitungen (Ph. Eur.) entsprechen.
Herstellung
Zur Herstellung der Urtinktur werden die frischen Zwiebeln fein zerkleinert (2 000) und 18 h lang in einem geschlossenen Gefäß stehen gelassen. 1 Teil der Pflanzenmasse wird mit 1,4 Teilen Ethanol 86 % (m/m) 10 bis 30 Tage lang mazeriert und anschließend abgepresst. Der Presssaft wird filtriert. Aus dem Filtrat werden flüssige Verdünnungen nach Vorschrift 3a hergestellt.
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,890 bis 0,935
Anmerkung zur Monographie:
Unter „Prüfung auf Reinheit“ wird die Prüfung auf das Aussehen der Lösung gestrichen, da nach Durchführung der erforderlichen Filtration oft Probleme mit der Einhaltung der Spezifikation auftraten, obwohl alle anderen Spezifikationen über die gesamte Laufzeit eingehalten werden konnten. Hierzu gehört auch die Prüfung auf freies Iod und Iodat, bei der auf die gleichen Substanzen geprüft wird.
Arsenum iodatum
Arsenum jodatum
| AsI3 | M r 455,6 |
Verwendet wird Arsen(III)-iodid, das mindestens 97,0 und höchstens 101,0 Prozent AsI3 enthält, berechnet auf die getrocknete Substanz.
Eigenschaften
Scharlachrote oder granatrote, glänzende Kristalle oder Blättchen; löslich in Wasser und Ethanol unter Bildung eines geringen kristallinen Niederschlags, schwer löslich in Ether
Prüfung auf Identität
- A.
-
Die Prüflösung (siehe „Prüfung auf Reinheit“) gibt die Identitätsreaktion auf Arsen (2.3.1) und die Identitätsreaktion a auf Iodid (2.3.1).
- B.
-
Schmelztemperatur (2.2.14): 140 bis 144 °C
Prüfung auf Reinheit
Prüflösung: 0,6 g Substanz werden in Wasser R zu 15 ml gelöst. Die Lösung wird filtriert.
Chlorid, Bromid: höchstens 500 ppm
2,5 ml Prüflösung werden mit 3 ml Ammoniak-Lösung R und 5 ml Silbernitrat-Lösung R 1 versetzt. Die Mischung wird geschüttelt, bis die überstehende Lösung klar ist, und filtriert. Das Filtrat wird unter Nachwaschen des Filters mit Wasser R zu 18 ml verdünnt und mit 2 ml Salpetersäure R versetzt. Nach 2 min darf die Mischung nicht stärker getrübt sein als eine gleichzeitig hergestellte Referenzlösung aus 10 ml Chlorid-Lösung (5 ppm Cl) R, 3 ml verdünnter Ammoniak-Lösung R 1, 5 ml Silbernitrat-Lösung R 1 und 2 ml Salpetersäure R.
Freies Iod, Iodat: 10 ml Prüflösung werden mit 2 ml Dichlormethan R ausgeschüttelt. Die organische Phase darf nicht stärker gefärbt sein als 2 ml einer Mischung von 2,0 ml Stammlösung Rot und 98,0 ml Salzsäure (10 g · l−1 HCl) (2.2.2, Methode I) (freies Iod). Nach Zusatz von 0,2 ml verdünnter Schwefelsäure R und erneutem Schütteln darf die organische Phase ebenfalls nicht stärker gefärbt sein als 2 ml einer Mischung von 2,0 ml Stammlösung Rot und 98,0 ml Salzsäure (10 g · l−1 HCl) (2.2.2, Methode I) (Iodat).
Kalium: höchstens 300 ppm
0,250 g Substanz werden in Wasser R zu 25,0 ml gelöst. Der Gehalt an Kalium wird mit Hilfe der Atomemissionsspektrometrie (2.2.22, Methode I) bei 766,5 nm, oder alternativ der Atomabsorptionsspektrometrie (2.2.23, Methode I) bei 766,5 nm, bestimmt. Zur Herstellung der Referenzlösungen werden 1,906 g Kaliumchlorid R (1,000 g K), das zuvor bei 100 bis 105 °C getrocknet wurde, in Wasser R zu 1000,0 ml gelöst und die Lösung entsprechend verdünnt.
Trocknungsverlust (2.2.32): höchstens 3,0 Prozent, mit 1,000 g Substanz durch 24 h langes Trocknen im Exsikkator über Molekularsieb R bestimmt
Gehaltsbestimmung
0,300 g Substanz werden in 50 ml Wasser R gelöst und nach Zusatz von 0,2 ml Methylorange-Mischindikator-Lösung R mit Kaliumhydroxid-Lösung (0,1 mol · l−1) bis zum Farbumschlag von Rotorange über Grün nach Blau titriert.
1 ml Kaliumhydroxid-Lösung (0,1 mol · l−1) entspricht 15,191 mg AsI3.
Arzneiformen
Die Lösung D2 enthält mindestens 0,95 und höchstens 1,05 Prozent AsI3.
Die 1. Dezimalverreibung enthält mindestens 9,5 und höchstens 10,5 Prozent AsI3.
Herstellung
Lösung D2 nach Vorschrift 5a unter Berücksichtigung des tatsächlichen Gehalts mit Ethanol 15 % (m/m)
Die Herstellung sollte unter Lichtausschluss erfolgen, bei Bedarf kann filtriert werden.
Die Lösung D2 ist unverzüglich weiterzuverarbeiten. Die folgenden Verdünnungen werden mit Ethanol 43 % (m/m) hergestellt.
Verreibungen nach Vorschrift 6
Die Triturationen D1 und D2 sind unverzüglich weiterzuverarbeiten.
Eigenschaften
Die Lösung D2 ist eine schwach hellgelbe, klare Flüssigkeit.
Die 1. Dezimalverreibung ist ein gelbrötliches Pulver.
Prüfung auf Identität
Die Lösung D2 beziehungsweise die 1. Dezimalverreibung gibt die Identitätsreaktion auf Arsen (2.3.1) und die Identitätsreaktion a auf Iodid (2.3.1). Prüflösung ist die Lösung D2, im Falle der 1. Dezimalverreibung werden 1 g mit 10 ml Wasser R versetzt, gerührt und filtriert. Das Filtrat ist die Prüflösung.
Prüfung auf Reinheit
Aussehen der Lösung: Die Lösung D2 muss klar (2.2.1) sein.
Relative Dichte (2.2.5): 0,983 bis 0,986
Gehaltsbestimmung
Die Bestimmung erfolgt wie unter „Gehaltsbestimmung“ bei der Substanz angegeben mit 10,0 g der Lösung D2 beziehungsweise 1,00 g der 1. Dezimalverreibung.
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
- –
-
Prüfung auf Identität: Bei der DC-Methode werden die HPTLC-Bedingungen ergänzt und die Beschreibung der Zonen im Text durch eine schematische Darstellung (DC-Kasten) ersetzt.
- –
-
Prüfung auf Reinheit: Der Trockenrückstand wird aufgrund der vorliegenden Chargendaten auf 0,50 % geändert.
Blatta orientalis
Verwendet wird die lebende gemeine Küchenschabe Blatta orientalis L.
Beschreibung
Die Tiere verbreiten einen süßlichen, in der Regel als widerlich empfundenen Geruch.
Die gemeine Küchenschabe wird 20 bis 28 mm lang. Die größeren Weibchen sind dunkel schwarzbraun, haben einen breiteren Hinterleib und tragen am Rücken schuppenartige Überreste von Flügeln sowie am Hinterende ein Paar seitlich stehender, 2 bis 3 mm langer, gegliederter Anhänge (Cerci). Die kleineren Männchen sind von kastanienbrauner Farbe. Sie haben gut ausgebildete Überflügel, die die letzten Hinterleibsegmente nicht decken. Zusätzlich zu den Cerci besitzen sie noch ein Paar kleine, ungegliederte, mehr zur Körpermitte hin gelegene Griffel (Styli).
Der Körper der Tiere ist dorsoventral abgeflacht, die kräftig bedornten Beine nehmen von vorn nach hinten an Länge zu. Die Facettenaugen der Männchen sind größer als die der Weibchen. Die Antennen sind länger als der Körper und mit zahlreichen Sinneshaaren besetzt. Die Mundwerkzeuge tragen gehäuft Tast- und Geschmacksorgane.
Prüfung auf Reinheit
Amerikanische Schabe: 26 bis 28 mm große, rotbraune Tiere mit rotgelbem Halsschild mit zwei dunkelbraunen Flecken und Flügeln, die bei beiden Geschlechtern länger als der Körper sind (Periplaneta americana L.), dürfen nicht verwendet werden.
Arzneiformen
Herstellung
1 Teil lebende Tiere wird in einem geeigneten Gefäß durch Zufügen einer ausreichenden Menge an Ethanol 94 % (m/m), jedoch nicht mehr als 5 Teilen, getötet. Nach Zusatz der gleichen Menge einer Mischung von 83 Teilen Ethanol 94 % (m/m) und 17 Teilen Wasser werden die Tiere zerkleinert. Der Ansatz wird mit so viel Teilen Ethanol 86 % (m/m) versetzt, dass sich in der Summe 10 Teile Arzneiträger ergeben. Flüssige Verdünnungen werden nach Vorschrift 4b hergestellt, wobei für die 2. und 3. Dezimalverdünnung Ethanol 86 % (m/m) verwendet wird.
Eigenschaften
Die Urtinktur ist eine blassgelbe bis gelbe Flüssigkeit mit unangenehmem Geruch.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: die Urtinktur
Referenzlösung: 10 mg Serin R und 10 mg Ephedrinhydrochlorid R werden in 10 ml einer Mischung gleicher Volumteile Methanol R und Wasser R gelöst.
Platte: DC-Platte mit Kieselgel R (5 bis 40 µm) [oder DC-Platte mit Kieselgel R (2 bis 10 µm)]
Fließmittel: Wasser R, Essigsäure 99 % R, 1-Butanol R (16:16:68 V/V/V)
Auftragen: 40 µl; [oder 10 µl] Untersuchungslösung und 10 µl [oder 2,5 µl] Referenzlösung; bandförmig 20 mm [oder 10 mm]
Laufstrecke: 15 cm [oder 6 cm]
Detektion: Nach Verdunsten des Fließmittels im Warmluftstrom wird die Platte mit einer Lösung von Ninhydrin R (10 g · l−1) in Ethanol 96 % R besprüht und 5 bis 10 min lang bei 105 bis 110 °C erhitzt. Die Auswertung erfolgt im Tageslicht.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
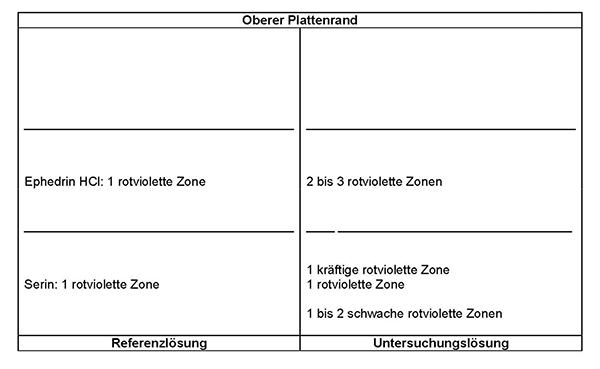
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,835 bis 0,855
Trockenrückstand (H 2.2.6): mindestens 0,50 Prozent
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Da bei der Gehaltsbestimmung der Symmetriefaktor für den Peak von Chelidonin oft außerhalb des üblichen Bereichs von 0,8–1,8 des Ph.-Eur.-Kapitels 2.2.46 lag, wird unter „Eignungsprüfung“ ein abweichender Symmetriefaktor von höchstens 2,0 aufgenommen.
Chelidonium majus
Chelidonium
Verwendet wird der frische Wurzelstock mit anhängenden Wurzeln von Chelidonium majus L. im Spätherbst oder zu Beginn des Austriebs.
Beschreibung
Der Wurzelstock hat einen schwach erdigen Geruch.
Die unterirdischen Organe bestehen aus einem vielköpfigen, orangebraunen bis dunkelbraunen, bis 30 mm dicken, 50 bis 100 mm langen Wurzelstock mit unregelmäßiger, borkiger bis tief längs gefurchter Oberfläche und einem mächtigen Wurzelsystem. Das weißliche, hellorangebraune oder dunkelbraune Gewebe des Wurzelstocks ist fein quer geringelt und von schwammig weicher Konsistenz. Er geht in eine ästig verzweigte Pfahlwurzel über, die mit vielen bis zu 5 mm dicken Seitenwurzeln besetzt ist. Die Pfahlwurzel ist unregelmäßig quer geringelt und matt orangebraun, die Nebenwurzeln sind heller orangebraun. Die äußere Rinde der Wurzel ist tiefgelb bis rotorange, der innere Teil und der deutlich abgesetzte Holzkörper sind heller gelborange. Aus dem Anschnitt von Wurzelstock und Wurzel tritt spontan oder beim Zusammendrücken ein tiefgelber bis oranger Milchsaft aus.
Arzneiformen
Die Urtinktur enthält mindestens 0,06 und höchstens 0,20 Prozent Alkaloide, berechnet als Chelidonin (C20H19NO5; Mr 353,4).
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 3a
Eigenschaften
Die Urtinktur ist eine braungelbe Flüssigkeit.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: 0,5 g Urtinktur werden mit 15 ml verdünnter Essigsäure R versetzt und 30 min lang im Wasserbad unter Umschwenken erwärmt. Nach dem Abkühlen wird die Mischung in einem Scheidetrichter mit 3 ml konzentrierter Ammoniak-Lösung R und 50 ml Chloroform R versetzt und 15 min lang kräftig geschüttelt. Die abgetrennte organische Phase wird unter vermindertem Druck zur Trockne eingeengt und der Rückstand in 1 ml Methanol R gelöst.
Referenzlösung: 10 mg Berberinchlorid R und 10 mg Chelidonin RH werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R
Fließmittel: Ameisensäure R, Wasser R, 1-Propanol R (1:9:90 V/V/V)
Auftragen: 40 µl Untersuchungslösung und 20 µl Referenzlösung; bandförmig (20 mm)
Laufstrecke: 15 cm
Detektion A: Nach Verdunsten des Fließmittels werden die Chromatogramme im ultravioletten Licht bei 365 nm ausgewertet.
Ergebnis A: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere fluoreszierende Zonen vorhanden sein.

Detektion B: Die Platte wird mit verdünntem Dragendorffs Reagenz R und danach mit Natriumnitrit-Lösung R besprüht. Die Auswertung erfolgt im Tageslicht.
Ergebnis B: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
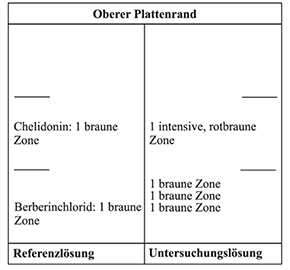
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,900 bis 0,920
Trockenrückstand (H 2.2.6): mindestens 1,2 Prozent
Gehaltsbestimmung
Flüssigchromatographie (2.2.29)
Untersuchungslösung: 0,60 g Urtinktur werden mit einer Mischung von 75 Volumteilen einer Lösung von Trifluoressigsäure R (1,5 ml · l–1) und 25 Volumteilen Methanol R 2 zu 5,0 ml verdünnt. Die Lösung wird über einen Filter der Porenweite 0,45 µm filtriert.
Stammlösung: 10,0 mg Chelidonin RH werden in Methanol R 2 zu 10,0 ml gelöst.
Referenzlösung: 2,0 ml Stammlösung werden mit einer Mischung von 75 Volumteilen einer Lösung von Trifluoressigsäure R (1,5 ml · l–1) und 25 Volumteilen Methanol R 2 zu 20,0 ml verdünnt.
Säule
- –
-
Größe: l = 0,15 m, Ø = 4,6 mm
- –
-
Stationäre Phase: nachsilanisiertes, alkyliertes Kieselgel zur Chromatographie zur Verwendung mit stark wässrigen mobilen Phasen R (4 μm)
- –
-
Temperatur: 20 °C
Mobile Phase
Mobile Phase A: eine Lösung von Trifluoressigsäure R (1,5 ml · l–1)
Mobile Phase B: Acetonitril zur Chromatographie R
| Zeit (min) |
Mobile Phase A (% V/V) |
Mobile Phase B (% V/V) |
|---|---|---|
| 0 – 5 | 73 | 27 |
| 5 – 18 | 73 → 40 | 27 → 60 |
| 18 – 19 | 40 → 0 | 60 → 100 |
| 19 – 20 | 0 → 73 | 100 → 27 |
| 20 – 27 | 73 | 27 |
Durchflussrate: 1,0 ml · min–1
Detektion: Spektrometer bei 291 nm
Einspritzen: 25 μl
Relative Retention (bezogen auf Chelidonin, tR etwa 11 min)
| – | Magnoflorin: | etwa 0,31 |
| – | Alkaloid 1: | etwa 0,40 |
| – | Alkaloid 2: | etwa 0,61 |
| – | Protopin: | etwa 0,81 |
| – | Allocryptopin: | etwa 0,90 |
| – | Coptisin: | etwa 1,10 |
| – | Alkaloid 3: | etwa 1,15 |
| – | Sanguinarin: | etwa 1,29 |
| – | Berberin: | etwa 1,38 |
| – | Chelerythrin: | etwa 1,52 |
Eignungsprüfung: Referenzlösung
- –
-
Wiederholpräzision: höchstens 2,0 Prozent relative Standardabweichung der Fläche des Chelidonin-Peaks nach 6 Einspritzungen
- –
-
Symmetriefaktor: höchstens 2,0 für den Peak von Chelidonin
Der Gehalt an Gesamtalkaloiden in Prozent (m/m), berechnet als Chelidonin, wird nach folgender Formel berechnet:
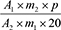
| A1 | = | Summe der Flächen aller Alkaloid-Peaks von Magnoflorin bis Chelerythrin im Chromatogramm der Untersuchungslösung |
| A2 | = | Fläche des Peaks von Chelidonin im Chromatogramm der Referenzlösung |
| m1 | = | Einwaage der Urtinktur in Gramm |
| m2 | = | Masse der Referenzsubstanz Chelidonin RH in Gramm |
| p | = | Prozentgehalt an Chelidonin in der Referenzsubstanz Chelidonin RH |
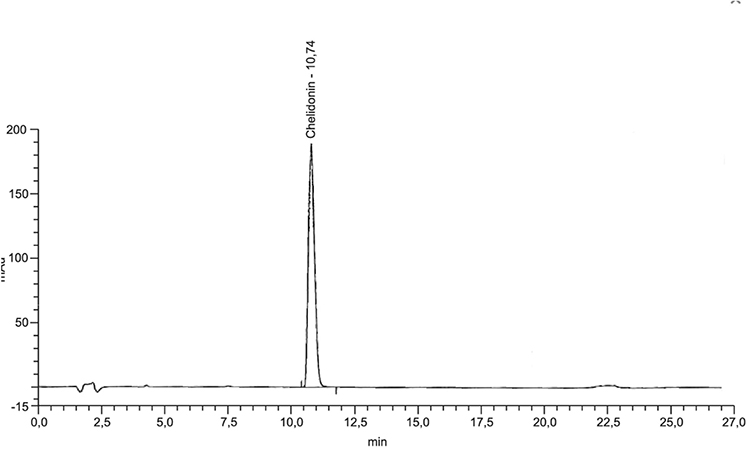
Beispielchromatogramm 1: Referenzlösung Chelidonium (0,1 mg · ml−1)
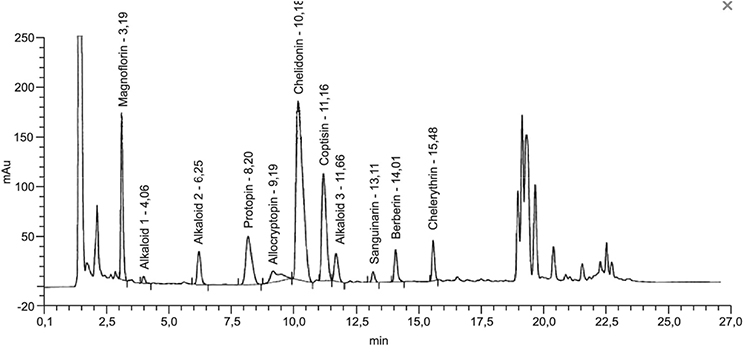
Beispielchromatogramm 2: Untersuchungslösung Chelidonium majus Ø, Vorschrift 3a
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Da bei der Gehaltsbestimmung der Symmetriefaktor für den Peak von Chelidonin oft außerhalb des üblichen Bereichs von 0,8–1,8 des Ph.-Eur.-Kapitels 2.2.46 lag, wird unter „Eignungsprüfung“ ein abweichender Symmetriefaktor von höchstens 2,0 aufgenommen.
Chelidonium majus e floribus, ethanol. Digestio
Chelidonium, Flos, ethanol. Digestio
Verwendet werden die frischen Blüten von Chelidonium majus L.
Beschreibung
Die gelben, radiären Blüten stehen in wenigblütigen, langgestielten, lockeren Dolden. Sie haben zwei blassgelbe, verstreut behaarte, hinfällige Kelchblätter, vier breit-eiförmige Kronblätter und zahlreiche gelbe Staubblätter. Der kurze, dicke Griffel hat eine zweilappige Narbe. Der längliche, aus zwei Fruchtblättern gebildete Fruchtknoten ist einfächerig und hat zahlreiche, zweireihig angeordnete Samenanlagen.
Arzneiformen
Die Urtinktur enthält mindestens 0,02 und höchstens 0,10 Prozent Alkaloide, berechnet als Chelidonin (C20H19NO5; Mr 353,4).
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 18c
Eigenschaften
Die Urtinktur ist eine braungelbe Flüssigkeit mit würzigem Geruch
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: 10 ml Urtinktur werden auf dem Wasserbad auf etwa 5 ml eingeengt. Der Rückstand wird mit 3 ml verdünnter Ammoniak-Lösung R 1 versetzt und die Mischung mit 10 ml Ether R ausgeschüttelt. Die abgetrennte organische Phase wird über wasserfreiem Natriumsulfat R getrocknet, dekantiert, zur Trockne eingedampft und der Rückstand in 1 ml Methanol R aufgenommen.
Referenzlösung: 10 mg Berberinchlorid R und 20 mg Methylrot R werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R
Fließmittel: Ameisensäure R, Wasser R, 1-Propanol R (1:9:90 V/V/V)
Auftragen: 40 µl Untersuchungslösung und 10 µl Referenzlösung; bandförmig (20 mm)
Laufstrecke: 10 cm
Detektion A: Nach Verdunsten des Fließmittels werden die Chromatogramme im ultravioletten Licht bei 365 nm ausgewertet.
Ergebnis A: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere fluoreszierende Zonen vorhanden sein.

Detektion B: Die Platte wird mit verdünntem Dragendorffs Reagenz R und danach mit Natriumnitrit-Lösung R besprüht. Die Auswertung erfolgt im Tageslicht.
Ergebnis B: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere braune oder braungelbe Zonen vorhanden sein.

Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,900 bis 0,915
Trockenrückstand (H 2.2.6): mindestens 1,4 Prozent
Gehaltsbestimmung
Flüssigchromatographie (2.2.29)
Untersuchungslösung: 1,00 g Urtinktur werden mit einer Mischung von 75 Volumteilen einer Lösung von Trifluoressigsäure R (1,5 ml · l–1) und 25 Volumteilen Methanol R 2 zu 5,0 ml verdünnt. Die Lösung wird über einen Filter der Porenweite 0,45 µm filtriert.
Stammlösung: 10,0 mg Chelidonin RH werden in Methanol R 2 zu 10,0 ml gelöst.
Referenzlösung: 2,0 ml Stammlösung werden mit einer Mischung von 75 Volumteilen einer Lösung von Trifluoressigsäure R (1,5 ml · l–1) und 25 Volumteilen Methanol R 2 zu 20,0 ml verdünnt.
Säule
- –
-
Größe: l = 0,15 m, Ø = 4,6 mm
- –
-
Stationäre Phase: nachsilanisiertes, alkyliertes Kieselgel zur Chromatographie zur Verwendung mit stark wässrigen mobilen Phasen R (4 μm)
- –
-
Temperatur: 20 °C
Mobile Phase
Mobile Phase A: eine Lösung von Trifluoressigsäure R (1,5 ml · l–1)
Mobile Phase B: Acetonitril zur Chromatographie R
| Zeit (min) |
Mobile Phase A (% V/V) |
Mobile Phase B (% V/V) |
|---|---|---|
| 0 – 5 | 73 | 27 |
| 5 – 18 | 73 → 40 | 27 → 60 |
| 18 – 19 | 40 → 0 | 60 → 100 |
| 19 – 20 | 0 → 73 | 100 → 27 |
| 20 – 27 | 73 | 27 |
Durchflussrate: 1,0 ml · min–1
Detektion: Spektrometer bei 291 nm
Einspritzen: 25 μl
Relative Retention (bezogen auf Chelidonin, tR etwa 11 min)
| – | Protopin: | etwa 0,81 |
| – | Alkaloid 1: | etwa 1,03 |
| – | Coptisin: | etwa 1,06 |
| – | Alkaloid 2: | etwa 1,13 |
| – | Alkaloid 3: | etwa 1,18 |
| – | Berberin: | etwa 1,31 |
Hinweis: Nicht alle aufgeführten Peaks müssen vorhanden sein und weitere Peaks können auftreten.
Eignungsprüfung: Referenzlösung
- –
-
Wiederholpräzision: höchstens 2,0 Prozent relative Standardabweichung der Fläche des Chelidonin-Peaks nach 6 Einspritzungen
- –
-
Symmetriefaktor: höchstens 2,0 für den Peak von Chelidonin
Der Gehalt an Gesamtalkaloiden in Prozent (m/m), berechnet als Chelidonin, wird nach folgender Formel berechnet:
| A1 | = | Summe der Flächen aller Alkaloid-Peaks mit einer relativen Retention von 0,8 bis 1,5 im Chromatogramm der Untersuchungslösung |
| A2 | = | Fläche des Peaks von Chelidonin im Chromatogramm der Referenzlösung |
| m1 | = | Einwaage der Urtinktur in Gramm |
| m2 | = | Masse der Referenzsubstanz Chelidonin RH in Gramm |
| p | = | Prozentgehalt an Chelidonin in der Referenzsubstanz Chelidonin RH |
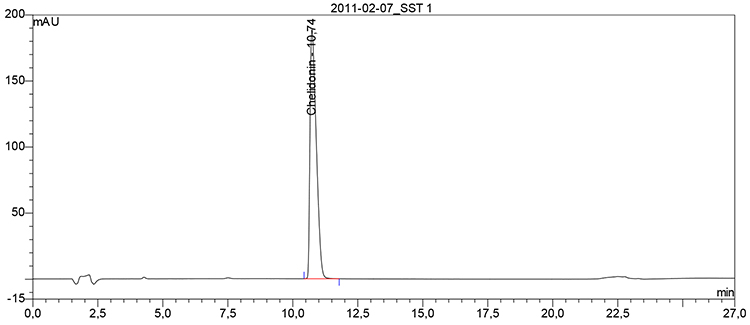
Beispielchromatogramm 1: Referenzlösung Chelidonin (0,1 mg ∙ ml–1)
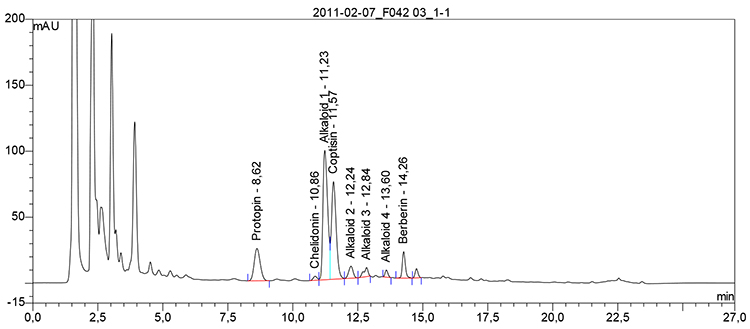
Beispielchromatogramm 2: Untersuchungslösung Chelidonium majus e floribus, ethanolische Digestio Ø, Vorschrift 18c
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Da bei der Gehaltsbestimmung der Symmetriefaktor für den Peak von Chelidonin oft außerhalb des üblichen Bereichs von 0,8–1,8 des Ph.-Eur.-Kapitels 2.2.46 lag, wird unter „Eignungsprüfung“ ein abweichender Symmetriefaktor von höchstens 2,0 aufgenommen.
Chelidonium majus Rh
Chelidonium Rh
Verwendet wird der frische Wurzelstock mit anhängenden Wurzeln von Chelidonium majus L. im Spätherbst oder zu Beginn des Austriebs.
Beschreibung
Der Wurzelstock hat einen schwach erdigen Geruch.
Die unterirdischen Organe bestehen aus einem vielköpfigen, orangebraunen bis dunkelbraunen, bis 30 mm dicken, 50 bis 100 mm langen Wurzelstock mit unregelmäßiger, borkiger bis tief längs gefurchter Oberfläche und einem mächtigen Wurzelsystem. Das weißliche, hell orangebraune oder dunkelbraune Gewebe des Wurzelstocks ist fein quer geringelt und von schwammig weicher Konsistenz. Er geht in eine ästig verzweigte Pfahlwurzel über, die mit vielen bis zu 5 mm dicken Seitenwurzeln besetzt ist. Die Pfahlwurzel ist unregelmäßig quer geringelt und matt orangebraun, die Nebenwurzeln sind heller orangebraun. Die äußere Rinde der Wurzel ist tiefgelb bis rotorange, der innere Teil und der deutlich abgesetzte Holzkörper sind heller gelborange. Aus dem Anschnitt von Wurzelstock und Wurzel tritt spontan oder beim Zusammendrücken ein tiefgelber bis orangefarbener Milchsaft aus.
Arzneiformen
Die Urtinktur enthält mindestens 0,10 und höchstens 0,50 Prozent Alkaloide, berechnet als Chelidonin (C20H19NO5; Mr 353,4).
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 21
Eigenschaften
Die Urtinktur ist eine braune Flüssigkeit.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: 0,5 g Urtinktur werden mit 15 ml verdünnter Essigsäure R versetzt und 30 min lang im Wasserbad unter Umschwenken erwärmt. Nach dem Abkühlen wird die Mischung in einem Scheidetrichter mit 3 ml konzentrierter Ammoniak-Lösung R und 50 ml Chloroform R versetzt und 15 min lang kräftig geschüttelt. Die abgetrennte organische Phase wird unter vermindertem Druck zur Trockne eingeengt und der Rückstand in 1 ml Methanol R gelöst.
Referenzlösung: 10 mg Berberinchlorid R und 10 mg Chelidonin RH werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R
Fließmittel: Ameisensäure R, Wasser R, 1-Propanol R (1:9:90 V/V/V)
Auftragen: 60 µl Untersuchungslösung und 10 µl Referenzlösung; bandförmig (20 mm)
Laufstrecke: 15 cm
Detektion A: Nach Verdunsten des Fließmittels werden die Chromatogramme im ultravioletten Licht bei 365 nm ausgewertet.
Ergebnis A: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere fluoreszierende Zonen vorhanden sein.

Detektion B: Die Platte wird mit verdünntem Dragendorffs Reagenz R und danach mit Natriumnitrit-Lösung R besprüht. Die Auswertung erfolgt im Tageslicht.
Ergebnis B: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung kann die braune Zone in Höhe der Referenzsubstanz Chelidonin auch sehr schwach sein. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
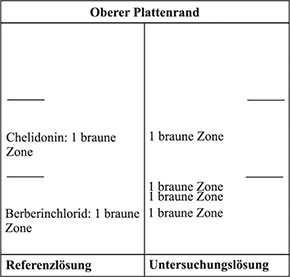
Prüfung auf Reinheit
Relative Dichte (2.2.5): 1,015 bis 1,055
Trockenrückstand (H 2.2.6): mindestens 3,0 Prozent
Gehaltsbestimmung
Flüssigchromatographie (2.2.29)
Untersuchungslösung: 0,60 g Urtinktur werden mit einer Mischung von 75 Volumteilen einer Lösung von Trifluoressigsäure R (1,5 ml ∙ l–1) und 25 Volumteilen Methanol R 2 zu 5,0 ml verdünnt. Die Lösung wird über einen Filter der Porenweite 0,45 µm filtriert.
Stammlösung: 10,0 mg Chelidonin RH werden in Methanol R 2 zu 10,0 ml gelöst.
Referenzlösung: 2,0 ml Stammlösung werden mit einer Mischung von 75 Volumteilen einer Lösung von Trifluoressigsäure R (1,5 ml ∙ l–1) und 25 Volumteilen Methanol R 2 zu 20,0 ml verdünnt.
Säule
- –
-
Größe: l = 0,15 m, Ø = 4,6 mm
- –
-
Stationäre Phase: nachsilanisiertes, alkyliertes Kieselgel zur Chromatographie zur Verwendung mit stark wässrigen mobilen Phasen R (4 μm)
- –
-
Temperatur: 20 °C
Mobile Phase
Mobile Phase A: eine Lösung von Trifluoressigsäure R (1,5 ml ∙ l–1)
Mobile Phase B: Acetonitril zur Chromatographie R
| Zeit (min) |
Mobile Phase A (% V/V) |
Mobile Phase B (% V/V) |
|---|---|---|
| 0 – 5 | 73 | 27 |
| 5 – 18 | 73 → 40 | 27 → 60 |
| 18 – 19 | 40 → 0 | 60 → 100 |
| 19 – 20 | 0 → 73 | 100 → 27 |
| 20 – 27 | 73 | 27 |
Durchflussrate: 1,0 ml · min–1
Detektion: Spektrometer bei 291 nm
Einspritzen: 25 μl
Relative Retention (bezogen auf Chelidonin, tR etwa 11 min)
| – | Alkaloid 1: | etwa 0,28 |
| – | Alkaloid 2: | etwa 0,38 |
| – | Alkaloid 3: | etwa 0,58 |
| – | Alkaloid 4: | etwa 0,61 |
| – | Protopin: | etwa 0,84 |
| – | Allocryptopin: | etwa 0,95 |
| – | Coptisin: | etwa 1,07 |
| – | Alkaloid 5: | etwa 1,10 |
| – | Sanguinarin: | etwa 1,20 |
| – | Berberin: | etwa 1,28 |
| – | Chelerythrin: | etwa 1,40 |
Hinweis: Nicht alle aufgeführten Peaks müssen vorhanden sein und weitere Peaks können auftreten.
Eignungsprüfung: Referenzlösung
- –
-
Wiederholpräzision: höchstens 2,0 Prozent relative Standardabweichung der Fläche des Chelidonin-Peaks nach 6 Einspritzungen
- –
-
Symmetriefaktor: höchstens 2,0 für den Peak von Chelidonin
Der Gehalt an Gesamtalkaloiden in Prozent (m/m), berechnet als Chelidonin, wird nach folgender Formel berechnet:
| A1 | = | Summe der Flächen aller Alkaloid-Peaks mit einer relativen Retention von 0,25 bis 1,60 im Chromatogramm der Untersuchungslösung |
| A2 | = | Fläche des Peaks von Chelidonin im Chromatogramm der Referenzlösung |
| m1 | = | Einwaage der Urtinktur in Gramm |
| m2 | = | Masse der Referenzsubstanz Chelidonin RH in Gramm |
| p | = | Prozentgehalt an Chelidonin in der Referenzsubstanz Chelidonin RH |
Beispielchromatogramm 1: Referenzlösung Chelidonin (0,1 mg ∙ ml–1)
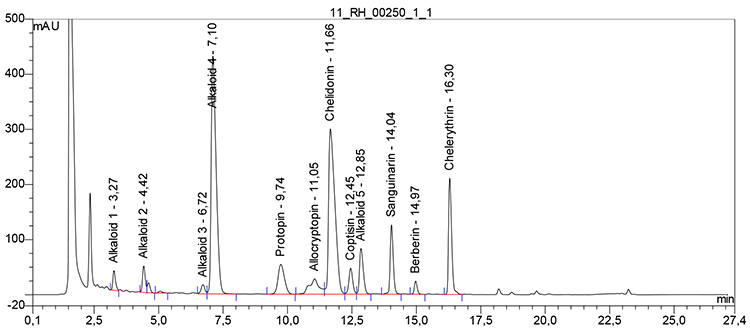
Beispielchromatogramm 2: Untersuchungslösung Chelidonium majus Rh Ø, Vorschrift 21
Lagerung
Dicht verschlossen, vor Licht geschützt
Anmerkungen zur Monographie:
Die Gehaltsbestimmungsmethode mittels Atomabsorptionsspektrometrie (AAS) auf Antimon wird aufgrund von Problemen mit der Reproduzierbarkeit durch eine bromatometrische Titration ersetzt.
Die gleiche Änderung wird in den HAB-Monographien „Stibium arsenicosum“, „Stibium sulfuratum aurantiacum (Antimonium sulfuratum aurantiacum)“ und „Stibium sulfuratum nigrum“ vorgenommen.
Hydrargyrum stibiato-sulfuratum
Aethiops antimonialis
Verwendet wird eine Verreibung aus gleichen Teilen Stibium sulfuratum nigrum und Hydrargyrum sulfuratum nigrum, die mindestens 46,6 und höchstens 52,8 Prozent Sb2S3 (Mr 339,7) sowie mindestens 22,5 und höchstens 27,7 Prozent Hg (Ar 200,6) enthält.
Herstellung
Gleiche Teile Stibium sulfuratum nigrum (HAB) und Hydrargyrum sulfuratum nigrum (HAB) werden 15 min lang intensiv miteinander verrieben.
Eigenschaften
Feines, schwarzes Pulver; praktisch unlöslich in Wasser, teilweise löslich in einer Mischung von 1 Volumteil Salpetersäure 65 % und 3 Volumteilen Salzsäure 36 %
Prüfung auf Identität
- A.
-
0,1 g Substanz werden mit 3 ml einer Mischung von 1 Volumteil Salpetersäure R und 3 Volumteilen Salzsäure R versetzt. Die Mischung wird kräftig geschüttelt und filtriert. Das Filtrat gibt die Identitätsreaktion a auf Quecksilber (2.3.1).
- B.
-
0,1 g Substanz werden mit 2 ml Salzsäure R versetzt. Die Mischung wird erhitzt und nach dem Abkühlen filtriert. Wird das Filtrat mit 1 ml einer Lösung von Molybdatophosphorsäure R (50 g · l−1) versetzt und die Mischung kurzzeitig erwärmt, färbt sie sich blau. Wird die Mischung mit Pentanol R ausgeschüttelt, färbt sich die organische Phase blau.
- C.
-
0,1 g Substanz werden mit 3 ml einer Mischung von 1 Volumteil Salpetersäure R und 3 Volumteilen Salzsäure R versetzt. Die Mischung wird so lange erwärmt, bis sich hellgelbe Abscheidungen zusammenballen.Anschließend wird die Mischung mit 3 ml Wasser R verdünnt und filtriert. Etwa ein Drittel des Rückstands wird mit 2 ml Piperidin R versetzt. Wird die Mischung 10 s lang geschüttelt, färbt sie sich allmählich rot.Zwei Drittel des Rückstands werden mit 2 ml Bromwasser R versetzt. Die Mischung wird erwärmt, bis sie sich entfärbt, filtriert und das Filtrat mit 5 ml Wasser R versetzt. Diese Lösung gibt die Identitätsreaktion a auf Sulfat (2.3.1).
Gehaltsbestimmung
A. Antimon als Sb2S3
0,100 g Substanz werden mit 20 ml Salzsäure R solange vorsichtig zum Sieden erhitzt, bis die entweichenden Dämpfe ein angefeuchtetes Blei(II)-acetat-Papier R nicht mehr verfärben. Es verbleibt ein unlöslicher Rückstand. Die abgekühlte Lösung wird mit 40 ml Wasser R verdünnt und mit Kaliumbromat-Lösung (0,0167 ∙ mol∙l–1) titriert. Gegebenenfalls muss noch Wasser R zugefügt werden, damit der Reaktionsbereich der Elektrode bedeckt ist.
Der Endpunkt wird mit Hilfe der Potentiometrie bestimmt (Ph. Eur., 2.2.20).
1 ml Kaliumbromat-Lösung (0,0167 mol · l−1) entspricht 8,49 mg Sb2S3.
B. Quecksilber
Die Bestimmung erfolgt mit Hilfe der Atomabsorptionsspektrometrie (2.2.23, Methode I).
Untersuchungslösung: 0,100 g Substanz werden in einem 100-ml-Messkolben unter Erwärmen in einer Mischung von 20 ml Salzsäure R und 3 ml Salpetersäure R gelöst.
Die Lösung wird nach dem Abkühlen mit Wasser R zu 100,0 ml verdünnt.
Referenzlösungen: Aus Quecksilber-Lösung (1 000 ppm) R werden mit verdünnter Salzsäure R Verdünnungen mit 200, 250 und 300 ppm Quecksilber hergestellt.
Die Absorption der Untersuchungslösung wird bei 253,7 nm mit einer Quecksilber-Hohlkathodenlampe als Strahlungsquelle bei einer spektralen Bandbreite von 0,5 nm und einer Luft-Acetylen-Flamme mit geeignetem Volumenverhältnis der Gase gemessen.
Arzneiformen
Die 1. Dezimalverreibung enthält mindestens 4,4 und höchstens 5,5 Prozent Sb2S3 sowie mindestens 2,1 und höchstens 2,9 Prozent Hg.
Herstellung
Verreibungen nach Vorschrift 6
Eigenschaften
Die 1. Dezimalverreibung ist ein graues Pulver.
Prüfung auf Identität
5,0 g der 1. Dezimalverreibung werden in 50 ml Wasser R suspendiert und die Mischung zentrifugiert. Die überstehende Lösung wird verworfen. Der Bodensatz wird in 20 ml Wasser R aufgeschüttelt und die Mischung erneut zentrifugiert. Der Rückstand gibt die Identitätsreaktionen der Substanz.
Gehaltsbestimmung
A. Antimon als Sb2S3
1,000 g der 1. Dezimalverreibung wird in einem Zentrifugenglas mit 20 ml einer Lösung, die 5 g Natriumchlorid R und 5 mg Natriumdodecylsulfat R in 100 ml Wasser R enthält, im Wasserbad suspendiert und die Mischung nach dem Abkühlen zentrifugiert. Die überstehende Flüssigkeit wird mittels Pipette abgehoben und verworfen. Der Vorgang wird 2-mal wiederholt. Die weitere Bestimmung erfolgt wie unter „Gehaltsbestimmung“ bei der Substanz angegeben.
B. Quecksilber
Die Bestimmung erfolgt wie unter „Gehaltsbestimmung“ bei der Substanz angegeben mit folgender Untersuchungslösung:
Untersuchungslösung: 1,00 g der 1. Dezimalverreibung wird in einem Zentrifugenglas mit 10 ml einer Lösung von 5 g wasserfreiem Natriumsulfat R und 5 mg Natriumdodecylsulfat R in 95 ml Wasser R versetzt und die Mischung geschüttelt. Die Suspension wird 5 min lang bei 4 000 U/min zentrifugiert und die überstehende Flüssigkeit verworfen. Der Vorgang wird 3-mal wiederholt. Der Rückstand wird unter schwachem Erwärmen auf dem Wasserbad in einem Teil einer Mischung von 20 ml Salzsäure R und 3 ml Salpetersäure R gelöst. Nach dem Abkühlen wird die Lösung mit der restlichen Säuremischung in einen 100-ml-Messkolben überführt und mit Wasser R zu 100,0 ml verdünnt.
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Die Gehaltsbestimmungsmethode mittels Atomabsorptionsspektrometrie (AAS) auf Arsen und Antimon wird aufgrund von Problemen mit der Reproduzierbarkeit durch eine iodometrische Titration von Arsen und eine bromatometrische Titration von Antimon ersetzt.
In den HAB-Monographien „Stibium sulfuratum aurantiacum (Antimonium sulfuratum aurantiacum)“, „Stibium sulfuratum nigrum“ und „Hydrargyrum stibiato-sulfuratum (Aethiops antimonialis)“ wird die Gehaltsbestimmung auf Antimon ebenfalls entsprechend geändert.
Stibium arsenicosum
Antimonium arsenicosum
Verwendet wird eine Mischung gleicher Teile Antimon(V)-oxid und Arsen(III)-oxid, die mindestens 47,5 und höchstens 52,5 Prozent Sb2O5 (Mr 323,5) sowie As2O3 (Mr 197,8) enthält.
Herstellung
Gleiche Teile Antimon(V)-oxid und Acidum arsenicosum (HAB) werden sorgfältig gemischt.
Eigenschaften
Gelblich weißes Pulver; schwer löslich in Wasser, teilweise löslich in Alkalihydroxid-Lösungen und warmer Salzsäure
Prüfung auf Identität
- A.
-
0,1 g Substanz werden mit 0,1 g Natriumcarbonat R und 2,5 ml Wasser R versetzt. Die Mischung wird erwärmt und filtriert. Das eventuell noch trübe Filtrat wird mit 1 ml Salzsäure R und 5 ml Hypophosphit-Reagenz R versetzt. Wird die Mischung 15 min lang auf dem Wasserbad erwärmt, entsteht ein schwarzer Niederschlag.
- B.
-
Das Filterpapier mit dem Rückstand der Identitätsreaktion A wird mit 10 ml Natriumcarbonat-Lösung R gewaschen. Wird das Papier mit einer Mischung von 1,5 ml Kaliumiodid-Lösung R und 2 ml Salzsäure R benetzt, entsteht eine sich innerhalb von 2 bis 3 min intensivierende braunrote bis rotbraune Färbung.
Prüfung auf Reinheit
Aussehen der Lösung: 0,50 g Substanz werden mit 5,0 ml verdünnter Ammoniak-Lösung R 1 versetzt. Die Mischung wird einige Minuten lang geschüttelt und anschließend zentrifugiert. Die überstehende Lösung muss farblos (2.2.2, Methode I) sein.
Sauer oder alkalisch reagierende Verunreinigungen: 0,50 g Substanz werden mit 10,0 ml Wasser R versetzt. Die Mischung wird 1 min lang geschüttelt und filtriert. Das schwach getrübte Filtrat wird mit 0,1 ml Methylorange-Lösung R versetzt. Bis zum Farbumschlag nach Gelb dürfen höchstens 0,5 ml Natriumhydroxid-Lösung (0,01 mol · l–1) und anschließend bis zum Farbumschlag nach Rot höchstens 0,75 ml Salzsäure (0,01 mol · l–1) verbraucht werden.
Gehaltsbestimmung
A. Arsen als As2O3
0,150 g Substanz werden in einem Zentrifugenglas mit 20 ml einer Mischung von 10 ml verdünnter Natriumhydroxid-Lösung R und 10 ml Wasser R suspendiert und die Mischung zentrifugiert. Die überstehende Flüssigkeit wird mittels Pipette abgehoben. Der Vorgang wird 2-mal wiederholt. Die vereinigten Zentrifugate werden mit 10 ml Salzsäure R1 versetzt und die Lösung mit 3 g Natriumhydrogencarbonat R neutralisiert. Nach Zusatz von 1 ml Stärke-Lösung R wird mit Iod-Lösung (0,05 mol · l–1) bis zur Blaufärbung titriert.
1 ml Iodlösung (0,05 mol · l–1) entspricht 4,946 mg As2O3.
B. Antimon als Sb2O5
Der im Zentrifugenglas verbliebene Rückstand wird portionsweise mit 20 ml Salzsäure R und mit Hilfe einer 3 ml-Kunststoffpipette in einen 100 ml Jodzahlkolben überführt und unter Rückflusskühlung bis zum Lösen der Substanz erhitzt. Nach dem Abkühlen werden 1 g Kaliumbromid R und 15 ml einer Lösung von Schwefeldioxid R (50 g ∙ l–1) in Wasser R4 zugegeben und anschließend 15 min offen erhitzt. Nach dem Abkühlen wird die Lösung mit 25 ml Wasser R verdünnt und mit Kaliumbromat-Lösung (0,0167 mol ∙ l–1) titriert.
Gegebenenfalls muss noch Wasser R zugefügt werden, damit der Reaktionsbereich der Elektrode bedeckt ist.
Der Endpunkt wird mit Hilfe der Potentiometrie bestimmt (Ph. Eur., 2.2.20).
1 ml Kaliumbromat-Lösung (0,0167 mol · l–1) entspricht 8,088 mg Sb2O5.
Arzneiformen
Die 1. Dezimalverreibung enthält mindestens 4,5 und höchstens 5,5 Prozent As2O3 sowie Sb2O5.
Herstellung
Verreibungen nach Vorschrift 6
Eigenschaften
Die 1. Dezimalverreibung ist ein fast weißes Pulver.
Prüfung auf Identität
1 g der 1. Dezimalverreibung gibt die Identitätsreaktionen der Substanz.
Gehaltsbestimmung
1,500 g der 1. Dezimalverreibung werden mit 20 ml einer Mischung von 10 ml verdünnter Natriumhydroxid-Lösung R und 10 ml Wasser R suspendiert und die Mischung zentrifugiert. Der Vorgang wird 2-mal wiederholt. Die weitere Bestimmung erfolgt wie unter „Gehaltsbestimmung“ bei der Substanz angegeben.
Anmerkungen zur Monographie:
Die Gehaltsbestimmungsmethode mittels Atomabsorptionsspektrometrie (AAS) auf Antimon wird aufgrund von Problemen mit der Reproduzierbarkeit durch eine bromatometrische Titration ersetzt.
Die gleiche Änderung wird in den HAB-Monographien „Stibium arsenicosum“, „Stibium sulfuratum nigrum“ und „Hydrargyrum stibiato-sulfuratum (Aethiops antimonialis)“ vorgenommen.
Stibium sulfuratum aurantiacum
Antimonium sulfuratum aurantiacum
Verwendet wird „Goldschwefel“, eine durch Fällung gewonnene Mischung von Antimon(V)-sulfid (Sb2S5) und Schwefel, die mindestens 52,0 und höchstens 68,0 Prozent Sb (Ar 121,8) enthält.
Eigenschaften
Feines, orangerotes Pulver mit schwachem Geruch nach Schwefelverbindungen; löslich in konzentrierter Salzsäure, Ammoniumsulfid-Lösung, Alkalisulfid-Lösungen und Alkalihydroxid-Lösungen
Prüfung auf Identität
- A.
-
50 mg Substanz werden in 2 ml verdünnter Natriumhydroxid-Lösung R unter Erwärmen gelöst. Wird die Lösung mit verdünnter Salzsäure R angesäuert, entsteht ein gelbroter Niederschlag.
- B.
-
0,1 g Substanz werden in einem Reagenzglas mit 2 ml Salzsäure R 1 versetzt. Wird die Mischung erwärmt, färben die entweichenden Dämpfe angefeuchtetes Blei(II)-acetat-Papier R schwarzbraun.
Prüfung auf Reinheit
Prüflösung: 0,10 g Substanz werden mit 50 ml Wasser R versetzt. Die Mischung wird 1 min lang geschüttelt und anschließend durch ein gehärtetes Filter filtriert.
Chlorid (2.4.4): 8,3 ml Prüflösung werden mit Wasser R zu 15 ml verdünnt. Diese Lösung muss der Grenzprüfung auf Chlorid entsprechen (0,3 Prozent).
Sulfat (2.4.13): 7,5 ml Prüflösung werden mit Wasser R zu 15 ml verdünnt. Diese Lösung muss der Grenzprüfung auf Sulfat entsprechen (1 Prozent).
Arsen (2.4.2): 0,5 g Substanz werden mit 10 ml Salpetersäure R versetzt. Die Mischung wird auf dem Wasserbad zur Trockne eingedampft, der Rückstand in 5 ml verdünnter Salzsäure R aufgenommen und die Mischung filtriert. 0,5 ml Filtrat müssen der Grenzprüfung B auf Arsen entsprechen (100 ppm).
In Salzsäure unlöslicher Glührückstand: höchstens 0,5 Prozent
5,000 g Substanz werden in 50 ml Salzsäure R unter Erwärmen gelöst. Die Lösung wird mit 50 ml Wasser R versetzt und die Mischung durch ein aschefreies Filter filtriert. Der Rückstand wird mit verdünnter Salzsäure R und Wasser R gewaschen, das Filter im Porzellantiegel verascht und der Rückstand bei etwa 800 °C bis zur Massekonstanz geglüht.
Gehaltsbestimmung
0,100 g Substanz werden mit 20 ml Salzsäure R solange vorsichtig zum Sieden erhitzt, bis die entweichenden Dämpfe ein angefeuchtetes Blei(II)-acetat-Papier R nicht mehr verfärben. Es erfolgt eine Abscheidung von Schwefel. Die abgekühlte Lösung wird mit 40 ml Wasser R verdünnt und mit Kaliumbromat-Lösung (0,0167 mol ∙ l–1) titriert. Gegebenenfalls muss noch Wasser R zugefügt werden, damit der Reaktionsbereich der Elektrode bedeckt ist.
Der Endpunkt wird mit Hilfe der Potentiometrie bestimmt (Ph. Eur., 2.2.20).
1 ml Kaliumbromat-Lösung (0,0167 mol · l–1) entspricht 6,09 mg Sb.
Arzneiformen
Die 1. Dezimalverreibung enthält eine mindestens 5,0 und höchstens 7,0 Prozent Sb entsprechende Menge Stibium sulfuratum aurantiacum.
Herstellung
Verreibungen nach Vorschrift 6
Eigenschaften
Die 1. Dezimalverreibung ist ein orangerotes Pulver.
Prüfung auf Identität
5 g der 1. Dezimalverreibung werden mit 50 ml Wasser R versetzt. Die Mischung wird unter gelegentlichem Umschwenken 1 h lang stehengelassen und anschließend filtriert. Der Rückstand wird zunächst mit Wasser R, anschließend mit Aceton R gewaschen und bei Raumtemperatur getrocknet. Er gibt die Identitätsreaktionen der Substanz.
Gehaltsbestimmung
1,000 g der 1. Dezimalverreibung wird in einem Zentrifugenglas mit 20 ml einer Lösung, die 5 g Natriumchlorid R und 5 mg Natriumdodecylsulfat R in 100 ml Wasser R enthält, im Wasserbad suspendiert und die Mischung nach dem Abkühlen zentrifugiert. Die überstehende Flüssigkeit wird mittels Pipette abgehoben und verworfen. Der Vorgang wird 2-mal wiederholt. Die weitere Bestimmung erfolgt wie unter „Gehaltsbestimmung“ bei der Substanz angegeben.
Lagerung
Dicht verschlossen, vor Licht geschützt
Anmerkungen zur Monographie:
Die Gehaltsbestimmungsmethode mittels Atomabsorptionsspektrometrie (AAS) auf Antimon wird aufgrund von Problemen mit der Reproduzierbarkeit durch eine bromatometrische Titration ersetzt.
Die gleiche Änderung wird in den HAB-Monographien „Stibium arsenicosum“, „Stibium sulfuratum aurantiacum (Antimonium sulfuratum aurantiacum)“ und „Hydrargyrum stibiato-sulfuratum (Aethiops antimonialis)“ vorgenommen.
Stibium sulfuratum nigrum
| Sb2S3 | M r 339,7 |
Verwendet wird Antimon(III)-sulfid, das mindestens 98,0 und höchstens 100,5 Prozent Sb2S3 enthält.
Eigenschaften
Grauschwarzes, glänzendes, schweres, sehr feines Pulver; löslich unter Erwärmen in Salzsäure
Prüfung auf Identität
0,1 g Substanz werden in 2 ml Salzsäure R unter Erwärmen gelöst. Wird die Mischung mit 5 ml Wasser R versetzt, färbt sie sich gelborange.
Prüfung auf Reinheit
In Salzsäure unlösliche Verunreinigungen: höchstens 1,0 Prozent
2,00 g Substanz werden mit 40 ml Salzsäure R versetzt. Die Mischung wird 10 min lang zum Sieden erhitzt und anschließend durch einen Glassintertiegel (16) (2.1.2) filtriert. Der Rückstand wird 3-mal mit je 10 ml verdünnter Salzsäure R gewaschen und der Tiegel mit dem Rückstand bei 105 bis 110 °C bis zur Massekonstanz getrocknet.
Arsen: 0,50 g Substanz werden mit 5 ml Ammoniumcarbonat-Lösung R versetzt. Die Mischung wird 2 min lang bei 50 bis 60 °C unter wiederholtem Schütteln stehengelassen und anschließend filtriert. Wird das Filtrat mit 2 ml Salzsäure R versetzt, darf sich innerhalb von 6 h keine gelbe, flockige Fällung bilden.
Kupfer: 0,10 g Substanz werden in 5 ml Salzsäure R gelöst. Wird die Lösung mit 8 ml konzentrierter Ammoniak-Lösung R versetzt, darf sich die Mischung nicht blau färben.
Gehaltsbestimmung
0,100 g Substanz werden mit 20 ml Salzsäure R solange vorsichtig zum Sieden erhitzt, bis die entweichenden Dämpfe ein angefeuchtetes Blei(II)-acetat-Papier R nicht mehr verfärben. Die abgekühlte Lösung wird mit 40 ml Wasser R verdünnt und mit Kaliumbromat-Lösung (0,0167 mol ∙ l–1) titriert. Gegebenenfalls muss noch Wasser R zugefügt werden, damit der Reaktionsbereich der Elektrode bedeckt ist.
Der Endpunkt wird mit Hilfe der Potentiometrie bestimmt (Ph. Eur., 2.2.20).
1 ml Kaliumbromat-Lösung (0,0167 mol · l−1) entspricht 8,49 mg Sb2S3.
Arzneiformen
Die 1. Dezimalverreibung enthält mindestens 9,3 und höchstens 10,6 Prozent Sb2S3.
Herstellung
Verreibungen nach Vorschrift 6
Eigenschaften
Die 1. Dezimalverreibung ist ein schwarzgraues Pulver.
Prüfung auf Identität
1 g der 1. Dezimalverreibung gibt die Identitätsreaktion der Substanz.
Gehaltsbestimmung
1,000 g der 1. Dezimalverreibung wird in einem Zentrifugenglas mit 20 ml einer Lösung, die 5 g Natriumchlorid R und 5 mg Natriumdodecylsulfat R in 100 ml Wasser R enthält, im Wasserbad suspendiert und die Mischung nach dem Abkühlen zentrifugiert. Die überstehende Flüssigkeit wird mittels Pipette abgehoben und verworfen. Der Vorgang wird 2-mal wiederholt. Die weitere Bestimmung erfolgt wie unter „Gehaltsbestimmung“ bei der Substanz angegeben.
- 1
- Extrelutsäule-NT20-Fertigsäule
- 2
- Extrelutsäule-NT20-Fertigsäule
- 3
- geeignetes wasserabweisendes Filter
- 4
- entspricht kommerziell erhältlicher schwefliger Säure
Heiße News an die Redaktion senden




Neueste Kommentare