
Bekanntmachung einer Mitteilung zum Deutschen Arzneibuch (Empfehlungen der Fachausschüsse der Deutschen Arzneibuch-Kommission)
Bundesinstitut
für Arzneimittel und Medizinprodukte
Bekanntmachung
einer Mitteilung
zum Deutschen Arzneibuch
(Empfehlungen der Fachausschüsse
der Deutschen Arzneibuch-Kommission)
Auf Grund des § 7 Absatz 5 der Geschäftsordnung für die Deutsche Arzneibuch-Kommission und deren Gremien vom 17. Juli 2009 (Bekanntmachung vom 8. Oktober 2009, BfArM-Internetseite) sind Empfehlungen der Fachausschüsse der Deutschen Arzneibuch-Kommission den betroffenen Fach- und Wirtschaftskreisen zur Kenntnis zu bringen.
Die Fachausschüsse der Deutschen Arzneibuch-Kommission haben die unten genannten Entwürfe revidierter Monographien für die Aufnahme in das Deutsche Arzneibuch empfohlen.
Dies wird hiermit zur Kenntnis gebracht.
Revidierte Monographien:
Aluminiumacetat-tartrat-Lösung
Campherspiritus
Cystein
Sojaöl, Partiell hydriertes
Zinkpaste
Zinkpaste, Weiche
Des Weiteren hat der Fachausschuss Pharmazeutische Biologie die Streichung der folgenden Monographie empfohlen, da sie mit dem Nachtrag 11. in das Europäische Arzneibuch aufgenommen wurde. Sie wird hiermit zur Kenntnis gebracht (Anlage).
Zu streichende Monographien
Rhabarbertrockenextrakt, Eingestellter
Stellungnahmen zu den Entwürfen für das Deutsche Arzneibuch sind bis spätestens 16. September 2024 einschließlich an die Geschäftsstelle der Arzneibuch-Kommissionen im Bundesinstitut für Arzneimittel und Medizinprodukte, Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn, zu richten.
45.-3660-37010-/24
Bundesinstitut
für Arzneimittel und Medizinprodukte
In Vertretung
Prof. Dr. Knöß
Anmerkungen zur Monographie
Schwermetalle: Die Prüfung 2.4.8 kann entfallen. Die Regeln zu Verunreinigungen durch Elemente in der EP-Monographie „Pharmazeutische Zubereitungen“ mit Verweis auf den Allgemeinen Text „5.20 Verunreinigung durch Elemente“ sind ausreichend.
Aluminiumacetat-tartrat-Lösung
Aluminii acetatis tartratis solutio
Definition
Aluminiumacetattartrat-Lösung enthält mindestens 1,30 und höchstens 1,45 Prozent (m/m) Al (Ar 26,98) und mindestens 5,3 und höchstens 6,3 Prozent (m/m) C2H4O2 (Mr 60,1).
Herstellung
| Aluminiumsulfat | 30,0 Teile |
| Essigsäure 99 % | 10,9 Teile |
| Calciumcarbonat | 13,5 Teile |
| Gereinigtes Wasser | 160,0 Teile |
| Weinsäure | nach Bedarf |
Die Zubereitung kann wie folgt hergestellt werden:
Das Aluminiumsulfat wird bei Raumtemperatur in 135 Teilen Gereinigtem Wasser gelöst und in diese Lösung das Calciumcarbonat unter ständigem Rühren eingetragen. Nach Beendigung der Gasentwicklung wird die Mischung der Essigsäure mit dem restlichen Wasser zugesetzt. Das Gemisch wird unter häufigem Rühren mindestens 3 Tage lang bedeckt stehengelassen, bis keine Gasentwicklung mehr zu beobachten ist und der Niederschlag sich abgesetzt hat. Anschließend wird filtriert; bei der Filtration ist die Anwendung von Vakuum zu vermeiden. In je 100 Teilen des Filtrats werden 3,5 Teile Weinsäure gelöst.
Bei der Herstellung können auch andere Methoden angewandt werden, unter der Voraussetzung, dass die gleiche Qualität wie mit der beschriebenen Methode erzielt wird.
Eigenschaften
Aussehen: Klare, farblose bis schwach gelbliche Flüssigkeit.
Geruch: Nach Essigsäure.
Prüfung auf Identität
- A.
-
Die Verdünnung von 1 ml Substanz mit 1 ml Wasser R gibt nach Zusatz von 0,1 ml verdünnter Ammoniak-Lösung R1 einen weißen, gallertartigen Niederschlag.
- B.
-
2 ml Substanz werden auf dem Wasserbad zur Trockne eingedampft. Der Rückstand gibt die Identitätsreaktion auf Acetyl (2.3.1).
- C.
-
Die Substanz gibt die Identitätsreaktion b auf Tartrat (2.3.1).
Prüfung auf Reinheit
Prüflösung: 2,5 g Substanz werden mit destilliertem Wasser R zu 50 ml verdünnt.
Aussehen der Substanz: Die Substanz muss klar (2.2.1) und darf nicht stärker gefärbt sein als die Farbvergleichslösung G5 (2.2.2, Methode II).
Relative Dichte (2.2.5): 1,044 bis 1,058.
Acidität:
- a)
-
10,0 ml Substanz müssen nach Zusatz von 20 ml Wasser R, 2,0 g Kaliumnatriumtartrat R und 1,0 ml Phenolphthalein-Lösung R1 mindestens 12,0 ml und dürfen höchstens 14,0 ml Natriumhydroxid-Lösung (1 mol · l−1) bis zur Rosafärbung verbrauchen.
- b)
-
5 ml Prüflösung dürfen sich nach Zusatz von 0,05 ml Methylorange-Lösung R nicht rot färben.
Hexacyanoferrat(II): 5 ml Prüflösung dürfen sich nach Zusatz von 0,1 ml Eisen(III)-chlorid-Lösung R2 innerhalb von 15 min nicht grün oder blau färben.
Sulfat (2.4.13): 1,0 ml Prüflösung wird mit destilliertem Wasser R zu 10,0 ml verdünnt. 6,0 ml dieser Lösung, mit destilliertem Wasser R zu 15 ml verdünnt, müssen der Grenzprüfung auf Sulfat entsprechen (0,5 Prozent).
Calcium (2.4.3): 1,0 ml Prüflösung wird mit destilliertem Wasser R zu 10,0 ml verdünnt. 8,0 ml dieser Lösung, mit destilliertem Wasser R zu 15 ml verdünnt, müssen der Grenzprüfung auf Calcium entsprechen (0,25 Prozent).
Eisen (2.4.9): 10 ml Prüflösung müssen der Grenzprüfung auf Eisen entsprechen (20 ppm).
Gehaltsbestimmung
Aluminium: 2,50 g Substanz werden in wasserfreiem Ethanol R zu 20,0 ml gelöst. Das Aluminium wird nach „Komplexometrische Titrationen“ (2.5.11) bestimmt.
1 ml Natriumedetat-Lösung (0,1 mol · l−1) entspricht 2,698 mg Al.
Essigsäure: 4,50 g Substanz werden nach dem Mischen mit 20 ml Wasser R und 10 ml Phosphorsäure 85 % R unter Konstanthalten des Volumens im Wasserdampfstrom so lange destilliert, bis 250 ml Destillat übergegangen sind. Als Vorlage werden 25,0 ml Natriumhydroxid-Lösung (0,5 mol · l−1) verwendet. Der Überschuss an Natriumhydroxid-Lösung wird nach Zusatz von 0,2 ml Phenolphthalein-Lösung R1 mit Salzsäure (0,5 mol · l−1) titriert.
1 ml Natriumhydroxid-Lösung (0,5 mol · l−1) entspricht 30,03 mg C2H4O2.
Lagerung
Gut verschlossen.
Hinweis
Anstelle von Aluminiumacetat-Lösung oder Essigsaurer Tonerde ist Aluminiumacetat-tartrat-Lösung abzugeben.
Anmerkungen zur Monographie:
Identitätsprüfung B – Die Prüfung nutzt problematische Chemikalien (Kaliumdichromat, Piperidin).
Die Identitätsprüfung B wird an die Identitätsprüfung C aus der EP Monographie Ethanol 96 % angepasst.
Höhersiedende Substanzen, Methanol, 2-Propanol – Die Prüfungen werden gestrichen.
Die DAB Monographie Campherspiritus beschreibt eine Zubereitung. Alle Bestandteile müssen den Anforderungen der entsprechenden Stoffmonographien des Europäischen Arzneibuches entsprechen. Höhere Alkohole, Methanol und 2-Propanol werden durch die Monographie Ethanol 96 % begrenzt. Eine erneute Prüfung auf diese Verunreinigungen ist nicht erforderlich.
Campherspiritus
Spiritus camphoratus
Definition
Campherspiritus enthält mindestens 9,5 und höchstens 10,5 Prozent (m/m) Campher oder Racemischen Campher, C10H16O (Mr 152,2).
Herstellung
| D-Campher oder Racemischer Campher | 1 Teil |
| Ethanol 90 % (V/V) | 7 Teile |
| Gereinigtes Wasser | 2 Teile |
Die Zubereitung kann wie folgt hergestellt werden:
Der D-Campher oder Racemische Campher wird in Ethanol 90 % (V/V) gelöst und das Gereinigte Wasser hinzugefügt.
Bei der Herstellung können auch andere Methoden angewandt werden, unter der Voraussetzung, dass die gleiche Qualität wie mit der beschriebenen Methode erzielt wird.
Eigenschaften
Aussehen: Klare, farblose Flüssigkeit.
Geruch: Stark nach Campher.
Prüfung auf Identität
- A.
-
Werden 10 ml Substanz von 20 °C unter Umschwenken in kleinen Anteilen mit etwa 6 ml Wasser R von 20 °C versetzt, scheidet sich ein weißer, kristalliner Niederschlag ab. Der Niederschlag wird in 30 ml Methanol R gelöst. Die Lösung wird mit 1,0 g Hydroxylaminhydrochlorid R sowie 1,0 g wasserfreiem Natriumacetat R versetzt und 2 h lang unter Rückfluss zum Sieden erhitzt. Nach dem Erkalten werden 100 ml Wasser R zugesetzt. Nach Abfiltrieren wird der Niederschlag mit 10 ml Wasser R gewaschen, aus 10 ml einer Mischung von 4 Volumteilen Ethanol 96 % R und 6 Volumteilen Wasser R umkristallisiert und im Vakuum getrocknet. Die Schmelztemperatur (2.2.14) der Kristalle liegt zwischen 118 und 121 °C.
- B.
-
Bei Wasserdampftemperatur werden durch Destillation von 10 ml Substanz etwa 5 ml Destillat abgetrennt. 0,1 ml des Destillates werden in einem Reagenzglas mit 1 ml einer Lösung von Kaliumpermanganat R (10 g · l−1) und 0,2 ml verdünnter Schwefelsäure R gemischt. Die Öffnung des Reagenzglases wird sofort mit einem Filterpapier bedeckt, das mit einer frisch hergestellten Lösung von 0,1 g Nitroprussidnatrium R und 0,5 g Piperazin-Hexahydrat R in 5 ml Wasser R getränkt ist. Nach wenigen Minuten entsteht auf dem Papier eine intensive Blaufärbung, die nach 10 bis 15 min verblasst.
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,880 bis 0,885.
Brechungsindex (2.2.6): 1,372 bis 1,374.
Gehaltsbestimmung
0,250 g Substanz werden mit 2-Propanol R zu 10,0 ml verdünnt. Die Absorption (2.2.25) wird im Maximum bei 290 nm gemessen. Der Gehalt an C10H16O wird mit Hilfe der spezifischen Absorption 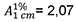 berechnet.
berechnet.
Lagerung
Dicht verschlossen.
Anmerkungen zur Monographie:
Eigenschaften: Geruch gestrichen
Identitätsprüfung B – Streichen der Probenvorbereitung (Harmonisierung mit Ph. Eur.)
Verwandte Substanzen – Stationäre Phase der DC aktualisiert. Angabe der DC-Laufstrecke als Verhältnis statt als absoluter Wert. (Harmonisierung mit Ph. Eur.)
Chlorid, Sulfat, Ammonium, Eisen: Redaktionelle Anpassung der Grenzprüfungen an den Stil der des Europäischen Arzneibuches.
Cystein
Cysteinum
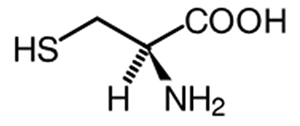
Definition
(R)-2-Amino-3-sulfanylpropansäure, C3H7NO2S, Mr 121,2.
Gehalt: 98,0 bis 101,0 Prozent (berechnet auf die getrocknete Substanz).
Eigenschaften
Aussehen: Weißes, kristallines Pulver oder farblose Kristalle.
Löslichkeit: Leicht löslich in Wasser und Ethanol, praktisch unlöslich in Ether.
Prüfung auf Identität
Die Prüfung B kann entfallen, wenn die Prüfungen A, C, D und E durchgeführt werden. Die Prüfungen C, D und E können entfallen, wenn die Prüfungen A und B durchgeführt werden.
- A.
-
Spezifische Drehung (siehe „Prüfung auf Reinheit“).
- B.
-
IR-Spektrum (2.2.24):Vergleich: Spektrum einer dem Arzneibuch entsprechenden Referenzsubstanz bekannter Identität.
- C.
-
2 ml einer Lösung der Substanz (5 g · l−1) werden mit Salzsäure (0,1 mol · l−1) auf einen pH-Wert von etwa 4 eingestellt, mit 0,5 ml einer Lösung von Ninhydrin R (2,5 g · l−1) versetzt und 10 min lang im Wasserbad erhitzt. Eine gelbe bis gelbbraune Färbung entsteht, die auf tropfenweisen Zusatz von 1 bis 2 ml Natriumhydroxid-Lösung (0,1 mol · l−1) zu der auf Raumtemperatur abgekühlten Lösung nach Rotviolett umschlägt.
- D.
-
5 ml einer Lösung der Substanz (1 g · l−1) werden mit 2 ml verdünnter Natriumhydroxid-Lösung R und 0,2 ml einer Lösung von Nitroprussidnatrium R (25 g · l−1) versetzt. Eine rote Färbung entsteht, die innerhalb von 2 min nach Gelb umschlägt.
- E.
-
0,10 g Substanz werden vorsichtig mit 1 ml Wasserstoffperoxid-Lösung 30 % R und 0,1 ml Eisen(III)-chlorid-Lösung R 1 versetzt. Nach dem Abkühlen auf Raumtemperatur werden 1 ml verdünnte Salzsäure R und 5 ml Wasser R zugegeben. Nach Zusatz von 1 ml Bariumchlorid-Lösung R 1 bildet sich innerhalb von 3 min eine weiße Trübung oder Fällung.
Prüfung auf Reinheit
Prüflösung: 2,5 g Substanz werden in kohlendioxidfreiem Wasser R zu 100 ml gelöst.
Die Prüflösung ist sofort nach der Herstellung zu verwenden.
Aussehen der Lösung: Die Prüflösung muss klar (2.2.1) und farblos (2.2.2, Methode II) sein.
pH-Wert (2.2.3): Zwischen 4,5 und 6,0, an der Prüflösung bestimmt.
Spezifische Drehung (2.2.7): +8,0 und +9,5, berechnet auf die getrocknete Substanz. Zur Bestimmung werden 3,00 g Substanz in verdünnter Salzsäure R zu 25,0 ml gelöst.
Verwandte Substanzen: Die Prüfung erfolgt mit Hilfe der Dünnschichtchromatographie (2.2.27).
Untersuchungslösung: 0,10 g Substanz werden in 6 ml Ethylmaleinimid-Lösung RN gelöst. Die Lösung wird zur Reaktion 15 min lang stehengelassen.
Referenzlösung: 1,0 ml Untersuchungslösung wird mit Wasser R zu 100 ml verdünnt.
Untersuchungsbedingungen
Stationäre Phase: DC-Platte mit Kieselgel R.
Auftragsvolumen: Je 5 µl, punktförmig, getrennt aufgetragen.
Fließmittel: Eine Mischung von 25 Volumteilen Wasser R, 25 Volumteilen Essigsäure 99 % R und 50 Volumteilen 1-ButanolR.
Laufstrecke: 2/3 der Platte.
Detektion und Auswertung
Die Chromatogramme werden 15 min lang bei 100 bis 105 °C getrocknet. Anschließend werden sie mit etwa 10 ml Ninhydrin-Lösung R (für eine 200-mm × 200-mm-Platte) besprüht und 10 min lang bei 100 bis 105 °C erhitzt. Kein im Chromatogramm der Untersuchungslösung auftretender Nebenfleck darf größer oder stärker gefärbt sein als der Fleck im Chromatogramm der Referenzlösung.
Chlorid (2.4.4): höchstens 400 ppm
5 ml Prüflösung werden auf 60 °C erhitzt und vorsichtig tropfenweise mit 5 ml Wasserstoffperoxid-Lösung 30 % R versetzt. Nach beendeter Zugabe wird diese Lösung 1 h lang zum Sieden erhitzt, auf Raumtemperatur abgekühlt und mit Wasser R zu 15 ml verdünnt.
Sulfat (2.4.13): höchstens 300 ppm
0,50 g Substanz werden in destilliertem Wasser R zu 15 ml gelöst.
Ammonium (N2.4.1): höchstens 200 ppm, mit 50 mg Substanz bestimmt.
Eisen (2.4.9): höchstens 20 ppm
In einem Scheidetrichter wird 0,50 g Substanz in 10 ml verdünnter Salzsäure R gelöst. Die Lösung wird 3-mal 3 min lang mit je 10 ml Isobutylmethylketon R 1 ausgeschüttelt. Die vereinigten organischen Phasen werden 3 min lang mit 10 ml Wasser R ausgeschüttelt. Die wässrige Phase wird verwendet.
Trocknungsverlust (2.2.32): Höchstens 0,5 Prozent, mit 1,000 g Substanz durch Trocknen im Trockenschrank bei 100 bis 105 °C bestimmt.
Sulfatasche (2.4.14): Höchstens 0,1 Prozent, mit 1,0 g Substanz bestimmt.
Gehaltsbestimmung
0,100 g Substanz werden in 10 ml verdünnter Salzsäure R gelöst und mit Wasser R zu 50 ml verdünnt. Nach dem Abkühlen in einem Eis-Wasser-Gemisch wird mit 2 g Kaliumiodid R und 15,0 ml Iod-Lösung (0,05 mol · l−1) versetzt und das verschlossene Reaktionsgefäß 15 min lang unter Lichtschutz stehengelassen. Mit Natriumthiosulfat-Lösung (0,1 mol · l−1) wird bis zur Entfärbung titriert, wobei gegen Ende der Titration 1 ml Stärke-Lösung R zugesetzt wird. Ein Blindversuch wird durchgeführt.
1 ml Iod-Lösung (0,05 mol · l−1) entspricht 12,12 mg C3H7NO2S.
Lagerung
Gut verschlossen, vor Licht geschützt.
Verunreinigungen, die durch diese Monographie begrenzt werden:
- A.
-
Cystin
- B.
-
Andere Aminosäuren.
Anmerkungen zur Monographie:
Streichung der Prüfung auf Nickel aufgrund ICH Q3D.
Streichung der Prüfungen auf Wasser und unverseifbare Anteile
Partiell hydriertes Sojaöl
Sojae oleum ex parte hydrogenatum
Definition
Partiell hydriertes Sojaöl wird durch teilweise Hydrierung von Sojaöl gewonnen.
Eigenschaften
Aussehen: Farblose bis gelbe, ölige Flüssigkeit bis weiße, fettartige Masse.
Geruch: Schwach, kakaoartig.
Löslichkeit: Praktisch unlöslich in Wasser und Ethanol, löslich in Toluol.
Prüfung auf Identität
- A.
-
Die Prüfung erfolgt nach „Identifizierung fetter Öle durch Dünnschichtchromatographie“ (2.3.2). Das erhaltene Chromatogramm entspricht dem Chromatogramm-Typ für Sojaöl.
- B.
-
Die Substanz entspricht der Prüfung „Iodzahl“ (siehe „Prüfung auf Reinheit“).
Prüfung auf Reinheit
Schmelztemperatur in der offenen Kapillare (2.2.15): Falls die Substanz fest oder halbfest ist, wird der Steigschmelzpunkt bestimmt.
Der Steigschmelzpunkt liegt in dem in der Beschriftung angegebenen Bereich. Die geschmolzene Substanz wird in die Kapillaren eingefüllt und 24 h lang bei 2 bis 10 °C stehengelassen.
Säurezahl (2.5.1): Höchstens 0,5, mit 10,00 g Substanz bestimmt. Die Substanz wird in 50 ml einer Mischung von gleichen Volumteilen Ethanol 96 % R und Toluol R unter Erwärmen gelöst. Die Prüfung wird mit der noch warmen Lösung durchgeführt.
Iodzahl (2.5.4): Die Iodzahl liegt in dem in der Beschriftung angegebenen Bereich. Peroxidzahl (2.5.5): Höchstens 5.
Alkalisch reagierende Substanzen: 2,00 g Substanz werden in einer Mischung von 1,5 ml Ethanol 96 % R und 3 ml Toluol R, falls notwendig unter leichtem Erwärmen, gelöst. Nach Zusatz von 0,05 ml Bromphenolblau-Lösung R dürfen bis zum Farbumschlag nach Gelb höchstens 0,4 ml Salzsäure (0,01 mol · l–1) verbraucht werden.
Lagerung
Vor Licht geschützt, in dicht verschlossenen, dem Verbrauch angemessenen, möglichst vollständig gefüllten Behältnissen.
Beschriftung
Die Beschriftung gibt insbesondere an
– falls die Substanz fest oder halbfest ist, den Bereich für den Steigschmelzpunkt
– den Bereich für die Iodzahl.
Anmerkungen zur Monographie:
Aktualisierung der Prüfung ‚Verhalten gegen Schwefelsäure‘, um Verwendung von Chromschwefelsäure zur Säuberung der Reagenzgläser zu vermeiden.
Zinkpaste
Zinci pasta
Definition
Zinkpaste enthält mindestens 23,5 und höchstens 26,5 Prozent Zinkoxid (ZnO, Mr 81,4) und 47 bis 53 Prozent Vaselin.
Herstellung
| Zinkoxid | 25 Teile |
| Weizenstärke | 25 Teile |
| Weißes Vaselin | 50 Teile |
Die Zubereitung kann wie folgt hergestellt werden:
Die Mischung von Zinkoxid und Weizenstärke wird in dünner Schicht 3 bis 4 h lang bei 40 bis 45 °C getrocknet, sofort gesiebt (250) und mit dem geschmolzenen Vaselin verrieben.
Bei der Herstellung können auch andere Methoden angewandt werden, unter der Voraussetzung, dass die gleiche Qualität wie mit der beschriebenen Methode erzielt wird.
Eigenschaften
Weiße, bei Raumtemperatur streichfähige, homogene Paste.
Geruch: Fast geruchlos.
Prüfung auf Identität
- A.
-
1,0 g Substanz wird mit 10 ml verdünnter Schwefelsäure R unter häufigem Umschütteln auf dem Wasserbad 5 min lang extrahiert. Nach dem Abkühlen wird filtriert. 1 ml Filtrat gibt nach Zusatz von 1 ml Kaliumhexacyanoferrat(II)-Lösung R einen weißen bis grünlich weißen Niederschlag.
- B.
-
0,5 ml des Filtrats von „Prüfung auf Identität“ A geben nach Zusatz von verdünnter Natriumhydroxid-Lösung R einen weißen, gallertartigen Niederschlag, der nach weiterem Zusatz wieder in Lösung geht.
- C.
-
Im toluolunlöslichen Rückstand unter „Farbe der Salbengrundlage“ ist Weizenstärke nachweisbar. Die Prüfung erfolgt unter dem Mikroskop unter Verwendung einer Mischung gleicher Volumteile Glycerol R und Wasser R. Die Droge zeigt große und kleine Körner und sehr selten Körner von mittlerer Größe. Die Großkörner von 10 bis 45 µm Durchmesser sind in der Flächenansicht scheibenförmig oder seltener nierenförmig. Spalt und Schichtungen sind nicht oder kaum sichtbar. Die Körner zeigen manchmal Risse in den Rändern. In der Seitenansicht sind die Körner elliptisch, spindelförmig und an der Längsachse aufgespalten. Die Kleinkörner sind rundlich oder polyedrisch und haben einen Durchmesser von 2 bis 10 µm. Im polarisierten Licht erscheint über dem Spalt ein ausgeprägtes Kreuz.
Prüfung auf Reinheit
Farbe der Salbengrundlage: 40 g Substanz werden unter Erwärmen mit 100 ml Toluol R geschüttelt. Die Suspension wird filtriert, das Filtrat auf dem Wasserbad eingedampft und der Rückstand 2 h lang bei 100 bis 105 °C getrocknet. 10 ml der flüssigen Schmelze dürfen nicht stärker gefärbt (2.2.2, Methode II) sein als eine Mischung von 1,0 ml Stammlösung Gelb und 9,0 ml Salzsäure 1 % RN.
Verhalten gegen Schwefelsäure: Ein Reagenzglas mit Schliffstopfen von etwa 125 mm Länge und etwa 18 mm innerem Durchmesser mit 2 Graduierungsmarken bei 5 und 10 ml wird mit heißem Wasser R (Temperatur mindestens 60 °C), Aceton R, Heptan R und anschließend mit Aceton R gewaschen, bei 100 bis 110 °C getrocknet und im Exsikkator erkalten gelassen. In dieses Reagenzglas werden 5 ml der unter „Farbe der Salbengrundlage“ erhaltenen flüssigen Schmelze und 5 ml Schwefelsäure 90 % RN 10 min lang im Wasserbad bei 70 °C erhitzt. Nach 5, 6 und 8 min wird das Reagenzglas jeweils für höchstens 3 s aus dem Wasserbad genommen und 3-mal kräftig nach unten geschlagen. Spätestens 5 min nach Beendigung des Erhitzens muss eine so weitgehende Trennung der Vaselin- und Schwefelsäure-Schicht erfolgt sein, dass ein Farbvergleich durchgeführt werden kann. Die Schwefelsäure-Schicht darf im durchfallenden Licht nicht stärker gefärbt (2.2.2, Methode I) sein als eine Mischung von 0,5 ml Stammlösung Blau, 1,5 ml Stammlösung Rot und 3,0 ml Stammlösung Gelb.
Gehaltsbestimmung Zinkoxid: 0,500 g Substanz werden in einem Weithalserlenmeyerkolben unter häufigem Umschwenken mit 25 ml verdünnter Salzsäure R so lange erwärmt, bis sich das Zinkoxid und die Weizenstärke vollständig gelöst haben und die Salbengrundlage klar auf der Lösung schwimmt. Nach Zusatz von 25 ml Toluol R wird mit Wasser R zu etwa 150 ml verdünnt und nach Zusatz von 0,1 ml Methylorange-Lösung R mit verdünnter Natriumhydroxid-Lösung R neutralisiert. Nach Zusatz von 10 ml Pufferlösung pH 10,9 R und 0,10 g Eriochromschwarz-T-Mischindikator RN wird mit Natriumedetat-Lösung (0,1 mol · l–1) bis zum Farbumschlag nach Grün titriert.
1 ml Natriumedetat-Lösung (0,1 mol · l–1) entspricht 8,14 mg ZnO.
Vaselin: 2,000 g Substanz werden mit 40 ml Toluol R unter Erwärmen digeriert. Die Mischung wird filtriert und der Rückstand 5-mal mit je 10 ml Toluol R gewaschen. Das Gesamtfiltrat wird auf dem Wasserbad eingedampft, der Rückstand 2 h lang bei 100 bis 105 °C getrocknet und gewogen.
Lagerung
Entspricht der Monographie Halbfeste Zubereitungen zur kutanen Anwendung (Praeparationes molles ad usum dermicum) der Ph. Eur. und folgender zusätzlicher Anforderung:
Vor Licht geschützt.
Beschriftung
Entspricht der Monographie Halbfeste Zubereitungen zur kutanen Anwendung.
Anmerkungen zur Monographie:
Streichung der Prüfung auf Blei und Cadmium aufgrund ICH Q3D.
Weiche Zinkpaste
Zinci pasta mollis
Definition
Weiche Zinkpaste enthält mindestens 28,0 und höchstens 32,0 Prozent Zinkoxid (ZnO, Mr 81,4).
Herstellung
| Zinkoxid | 30 Teile |
| Dickflüssiges Paraffin | 40 Teile |
| Weißes Vaselin | 20 Teile |
| Gebleichtes Wachs | 10 Teile |
Die Zubereitung kann wie folgt hergestellt werden: Das Zinkoxid (250) wird mit dem Dickflüssigen Paraffin zu einer gleichmäßigen Suspension verarbeitet. Die Suspension wird auf dem Wasserbad mit den übrigen Komponenten bis zum vollständigen Schmelzen erwärmt, gemischt und bis zum Erkalten gerührt. Bei der Herstellung können auch andere Methoden angewandt werden, unter der Voraussetzung, dass die gleiche Qualität wie mit der beschriebenen Methode erzielt wird.
Eigenschaften
Weiße, weiche Paste.
Geruch: Schwach nach Bienenwachs.
Prüfung auf Identität
- A.
-
2,5 g Substanz werden 5 min lang mit 25 ml Toluol R verrührt. Die Suspension wird über einen Glassintertiegel (16) filtriert und scharf abgesaugt. Der im Glassintertiegel verbleibende Rückstand wird mit etwa 20 ml Ether R fettfrei gewaschen und trockengesaugt. Der Rückstand wird in 15 ml verdünnter Salzsäure R unter Erwärmen auf dem Wasserbad gelöst und die Lösung mit Wasser R zu 25 ml verdünnt. Sie gibt die Identitätsreaktion auf Zink (2.3.1).
- B.
-
Die Prüfung erfolgt mit Hilfe der Dünnschichtchromatographie (2.2.27). Untersuchungslösung: 40 mg Substanz werden in 2 ml Dichlormethan R suspendiert. Referenzlösungen: Je 20 mg flüssiges Paraffin R, weißes Vaselin R, gebleichtes Wachs RN, mittelkettige Triglyceride RN und Wollwachsalkohole RN werden getrennt in jeweils 2 ml Dichlormethan R gelöst.
Untersuchungsbedingungen
Stationäre Phase: Kieselgel GF254R.
Auftragsvolumen: 10 µl Untersuchungslösung und 10 µl jeder Referenzlösung, bandförmig, getrennt aufgetragen.
Zur Nachaktivierung wird die Platte nach dem Auftragen der Lösungen 15 min lang an der Luft liegen gelassen.
Fließmittel: Eine Mischung von 1 Volumteil wasserfreier Essigsäure R, 10 Volumteilen Ether R und 90 Volumteilen Hexan R.
Laufstrecke: 15 cm.
Detektion und Auswertung
Die Chromatogramme werden mit einer Lösung von Ammoniumanilinonaphthalinsulfonat RN (1 g · l –1) besprüht. Die noch feuchte Platte wird im ultravioletten Licht bei 365 nm ausgewertet. Im oberen Rf-Bereich befindet sich im Chromatogramm der Untersuchungslösung die Zone von Vaselin und flüssigem Paraffin; gebleichtes Wachs lässt sich durch die Vielzahl der Einzelzonen identifizieren. Im Chromatogramm der Untersuchungslösung darf die Zone der mittelkettigen Triglyceride nicht auftreten. Zur Unterscheidung von anderen Rezepturen können auch die Zonen der Wollwachsalkohole hilfreich sein.
Gehaltsbestimmung
0,500 g Substanz werden in einem Weithalserlenmeyerkolben auf dem Wasserbad unter häufigem Umschwenken mit 25 ml verdünnter Salzsäure R so lange erwärmt, bis sich das Zinkoxid vollständig gelöst hat und die Salbengrundlage klar auf der Lösung schwimmt. Nach Zusatz von 25 ml Toluol R wird die Mischung mit Wasser R zu etwa 150 ml verdünnt und nach Zusatz von 0,1 ml Methylorange-Lösung R mit verdünnter Natriumhydroxid-Lösung R neutralisiert. Nach Zusatz von 10 ml Pufferlösung pH 10,9 R und 0,10 g Eriochromschwarz-T-Mischindikator RN wird mit Natriumedetat-Lösung (0,1 mol · l–1) bis zum Farbumschlag nach Grün titriert. 1 ml Natriumedetat-Lösung (0,1 mol · l–1) entspricht 8,14 mg ZnO.
Lagerung
Entspricht der Monographie Halbfeste Zubereitungen zur kutanen Anwendung (Praeparationes molles ad usum dermicum) der Ph. Eur. und folgender zusätzlicher Anforderung:
Vor Licht geschützt.
Beschriftung
Entspricht der Monographie Halbfeste Zubereitungen zur kutanen Anwendung.
Heiße News an die Redaktion senden




Neueste Kommentare