
Bundesinstitut
für Arzneimittel und Medizinprodukte
Bekanntmachung
einer Mitteilung zum Homöopathischen Arzneibuch
(Empfehlungen der Fachausschüsse
der Deutschen Homöopathischen Arzneibuch-Kommission)
Auf Grund des § 7 Absatz 5 der Geschäftsordnung für die Deutsche Homöopathische Arzneibuch-Kommission und deren Gremien (vgl. Bekanntmachung vom 17. Juli 2009, BfArM-Internetseite) sind Empfehlungen der Fachausschüsse der Deutschen Homöopathischen Arzneibuch-Kommission den betroffenen Fach- und Wirtschaftskreisen zur Kenntnis zu bringen.
Der Fachausschuss Analytik der Deutschen Homöopathischen Arzneibuch-Kommission hat neue Monographien und eine neue Reagenzienbeschreibung erarbeitet sowie Monographien überarbeitet.
Die Entwürfe sollen in das Homöopathische Arzneibuch aufgenommen werden, sie werden hiermit zur Kenntnis gebracht (Anlage).
Neue Monographien:
- 1.
-
Candida albicans e volumine cellulae (lyophil., steril.)
- 2.
-
Equisetum hiemale (Equisetum hyemale)
- 3.
-
Mucor racemosus e volumine cellulae (lyophil., steril.)
- 4.
-
Penicillium chrysogenum e volumine cellulae (lyophil., steril.)
Revidierte Monographien:
- 1.
-
Acidum formicicum
- 2.
-
Acorus calamus (Calamus aromaticus)
- 3.
-
Ambra grisea (Ambra)
- 4.
-
Aspergillus niger e volumine cellulae (lyophil., steril.)
- 5.
-
Euphrasia 3c (Euphrasia officinalis 3c)
- 6.
-
Ginkgo biloba (Ginkgo)
- 7.
-
Hydrargyrum nitricum oxydulatum (Mercurius nitricus oxydulatus)
- 8.
-
Solanum dulcamara (Dulcamara)
- 9.
-
Solanum nigrum
Neue Reagenzienbeschreibung:
- 1.
-
Phosphat-Pufferlösung pH 2,0 (0,2 mol · l–1) RH
Stellungnahmen zu den oben genannten Entwürfen des Homöopathischen Arzneibuches sind bis spätestens 28. Dezember 2021 einschließlich an die Geschäftsstelle der Arzneibuch-Kommissionen im Bundesinstitut für Arzneimittel und Medizinprodukte, Kurt-Georg-Kiesinger-Allee 3, 53175 Bonn, zu richten.
Bonn, den 5. Oktober 2021
43.07-2021-
Bundesinstitut
für Arzneimittel und Medizinprodukte
Prof. Dr. K. Broich
Neue Monographien
Candida albicans e volumine cellulae (lyophil., steril.)
Verwendet wird ein Lyophilisat, welches nach der Fermentation von Candida albicans (Robin) Berkhout 1923 aus der vom Kulturmedium befreiten Biomasse hergestellt wird, mit einem Gehalt von mindestens 100 mg bis höchstens 700 mg Gesamtprotein pro Gramm Lyophilisat.
Herstellung
Die Biomasse wird nach den Grundprinzipien der Monographie Fermentationsprodukte (Ph. Eur.) gewonnen. Als Kulturmedium wird eine Mischung von Glucose-Monohydrat (10,0 g · l–1), Caseinhydrolysat (5,0 g · l–1), Natriumchlorid (5,0 g · l–1), Ammoniumsulfat (5,0 g · l–1), Magnesiumsulfat-Heptahydrat (0,2 g · l–1), Biotin (0,001 g · l–1) und Kaliummonohydrogenphosphat (2,5 g · l–1) in Gereinigtem Wasser verwendet. Die Fermentation erfolgt unter Zufuhr von Luft bei 35 °C bis 39 °C und einem pH-Wert von 5,5 bis 7,5. Die Ernte erfolgt am Ende der stammspezifischen Wachstumsphase zu Beginn der Abflachung der Turbidimetrie-Kurve. Die Biomasse wird durch Zentrifugation von dem Kulturmedium getrennt. Danach wird im gleichen Verfahren die Biomasse mit Gereinigtem Wasser versetzt, gewaschen und ebenfalls durch Zentrifugation abgetrennt. Das Waschvolumen muss mindestens dem 10-fachen des Produktvolumens nach Zellernte entsprechen. Anschließend werden die Zellen in einer geeigneten Zellmühle mechanisch aufgeschlossen. Diese Phase wird von den wasserunlöslichen Bestandteilen zunächst durch Zentrifugation und anschließend durch eine Mikrofiltration (0,2 μm) im Querstrom-Verfahren getrennt. Das resultierende Filtrat wird anschließend einer Sterilfiltration (0,2 μm) im Durchlauf-Verfahren unterzogen und muss der Prüfung auf Sterilität (2.6.1) entsprechen. Anschließend wird die Lösung gefriergetrocknet. Dieses Lyophilisat besteht aus wasserlöslichen Zellbestandteilen und wasserunlöslichen Zellbestandteilen, die kleiner als 0,2 μm sind.
Beschreibung
Makroskopische Merkmale: Die vegetative Form der Hefe zeigt runde, schwach glänzende, weiße bis cremefarbene glattrandige Kolonien.
Mikroskopische Merkmale: Rundliche bis eiförmige Hefezellen, die echte Mycelien sowie Pseudomycelien bilden. Begrenzte Entwicklung von asexuellen Sporen
Lyophilisat: Gelbbraunes, amorphes Pulver
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (2.2.31): unter Verwendung eines 4- bis 20-prozentigen Gradientengels
Die SDS-PAGE kann unter Verwendung von kommerziell erhältlichen Gradientengelen durchgeführt werden.
Gele mit Gradienten (Trenngele) enthalten eine von oben nach unten zunehmende Konzentration an Acrylamid. Die Herstellung von Gradientengelen erfolgt mit einer gradientenbildenden Apparatur.
Ein 4 bis 20 Prozent Acrylamid enthaltenes Gradientengel wird hergestellt.
Eine 4-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
10 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
72 Volumteilen Wasser R
- –
-
2 Volumteilen einer Lösung von Natriumdodecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Eine 20-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
50 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
15 Volumteilen Wasser R
- –
-
17 Volumteilen einer Lösung von Saccharose R (855,8 g · l–1)
- –
-
2 Volumteilen einer Lösung von Natriumdodecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Die Kammern der gradientenbildenden Apparatur werden mit den Acrylamid-Lösungen beladen. Das polymerisierte Gradientengel wird gemäß den Geräteangaben hergestellt. Nach vollständiger Polymerisation wird das Gradientengel mit Wasser R gespült. Überschüssige Flüssigkeit wird entfernt. Die Lösung, die die Grundlage für das Anreicherungsgel bildet, wird in die Apparatur eingefüllt und ein sauberer Kamm wird eingelegt. Anschließend lässt man die Lösung des Anreicherungsgels polymerisieren.
Geldimension: 8,6 x 6,7 x 0,1 cm (Breite x Länge x Dicke)
SDS-PAGE-Laufpuffer (10x): 144 g Glycin R, 30 g Tris-Hydrochlorid (=Tris(hydroxymethyl)-aminomethan- bzw. Trometamolhydrochlorid) und 10 g Natriumdodecylsulfat R werden in Wasser R zu 1 000 ml gelöst. Der pH-Wert der Lösung sollte zwischen 8,1 und 8,8 liegen.
SDS-PAGE-Laufpuffer (1x): Vor Gebrauch wird der SDS-PAGE-Laufpuffer (10x) 1 zu 10 mit Wasser R verdünnt.
Tris-Hydrochlorid-Lösung (0,5 mol · l–1): 7,88 g Tris-Hydrochlorid wird mit Wasser R zu 100 ml verdünnt.
Natriumdodecylsulfat-Lösung (10 %): 5 g Natriumdodecylsulfat R werden mit Wasser R zu 50 ml verdünnt.
Bromphenolblau-Lösung (1 %): 0,05 g Bromphenolblau R werden mit Wasser R zu 5 ml verdünnt.
Proben-Pufferlösung: 20 ml Tris-Hydrochlorid-Lösung (0,5 mol · l–1), 16 ml Glycerol 85 % R 1, 32 ml Natriumdodecylsulfat-Lösung (10 %), 8 ml β-Mercaptoethanol R, 4 ml Bromphenolblau-Lösung (1 %) und 40 ml Wasser R werden gemischt.
Stammlösungen: Aus dem Lyophilisat wird eine Lösung hergestellt, die ungefähr 10 mg · ml–1 des Ausgangsmaterials enthält. Es können zusätzliche Verdünnungen wie 1 zu 2 oder 1 zu 5 hergestellt werden.
Untersuchungslösung: 150 µl der entsprechenden Stammlösung werden mit 50 µl Proben-Pufferlösung gemischt (beispielsweise in einem 1,5-ml-Eppendorf-Gefäß). Das Reaktionsgefäß wird verschlossen, in einem Wasserbad 5 min lang bei 95 °C erhitzt und danach auf Raumtemperatur abgekühlt.
Referenzlösung: Eine Lösung geeigneter Marker für Molekülmassen zur Kalibrierung von SDS-Polyacrylamidgelen im Bereich von 10 bis 250 kDa wird verwendet.
Detektion: Silberfärbung
Fixierlösung: 60 ml wasserfreies Ethanol R werden mit 20 ml wasserfreier Essigsäure R versetzt. Die Mischung wird mit 120 ml Wasser R verdünnt.
Silber-Färbelösung: 2 g Silbernitrat R und 0,2 ml Formaldehyd-Lösung R werden mit Wasser R zu 1 000 ml verdünnt.
Reduktionslösung: 314 mg Natriumthiosulfat R werden mit Wasser R zu 1 000 ml verdünnt.
Entwicklerlösung: 30 g wasserfreies Natriumcarbonat R, 0,5 ml Formaldehyd-Lösung R und 0,1 ml Reduktionslösung werden in Wasser R zu 1 000 ml verdünnt.
Blockierlösung: 5 g Glycin R werden in Wasser R zu 1 000 ml verdünnt.
Die Kassette mit dem Gel wird in die Elektrophorese-Apparatur eingebracht. Danach werden 600 ml des SDS-PAGE-Laufpuffers in die inneren und äußeren Kammern gefüllt. Dabei ist darauf zu achten, dass keine Luftblasen entstehen. Wenn notwendig, muss Puffer nachgefüllt werden. Danach wird der Kamm aus den Geltaschen entfernt und die Taschen mit SDS-PAGE-Laufpuffer gespült, um Luftbläschen oder nicht polymerisierte Acrylamid-Rückstände zu entfernen.
Auftragen: 10 µl Untersuchungslösung und 10 µl Referenzlösung
Laufbedingungen: Die Elektrophorese wird etwa 60 min lang bei Raumtemperatur unter Anlegen von 200 V durchgeführt. Diese Bedingungen gelten bei Verwendung eines Gels im oben genannten Format. Die Elektrophorese wird beendet, sobald die Farbstofffront die Referenzlinie auf der Kassette erreicht.
Der Lauf wird gestartet.
Silberfärbung: Nach dem Lauf wird das Gel aus der Kammer entfernt. Gegebenenfalls wird die Anleitung des Herstellers beachtet.
Folgende Schritte werden ausgeführt, wobei nach den Behandlungen die Lösungen jeweils vorsichtig abdekantiert werden:
- –
-
Fixieren: Das Gel wird 60 min lang unter leichtem Schütteln in Fixierlösung getaucht.
- –
-
Waschen: Das Gel wird 2-mal 20 min lang unter leichtem Schütteln in einer Mischung von 70 ml Wasser R und 30 ml wasserfreiem Ethanol R gewaschen.
- –
-
Waschen: Das Gel wird einmal 20 min lang unter leichtem Schütteln in Wasser R gewaschen.
- –
-
Reduktion: Das Gel wird etwa 1 min lang unter leichtem Schütteln in 100 ml Reduktionslösung getaucht.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Färben: Das Gel wird maximal 20 min lang unter leichtem Schütteln in Silber-Färbelösung getaucht. Das Gel bleibt nur so lange in der Färbelösung, bis sich eindeutige Banden ergeben. Danach kann der Färbeschritt beendet werden, auch wenn 20 min noch nicht abgelaufen sind.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Entwicklung: Das Gel wird etwa 3 bis 5 min lang unter leichtem Schütteln in 100 ml Entwicklerlösung getaucht.
- –
-
Waschen: Das Gel wird etwa 20 s lang unter leichtem Schütteln mit 100 ml Wasser R gewaschen.
- –
-
Blockieren: Das Gel wird etwa 10 min lang in Blockierlösung getaucht.
Ergebnis: Der Bereich der Molekülmassen und die Zonenfolge im Elektropherogramm der Untersuchungslösung sind aus den nachstehenden Angaben ersichtlich. Weitere Zonen können vorhanden sein.
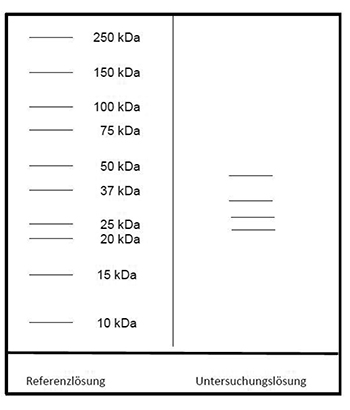
Prüfung auf Reinheit
Polyacrylamid-Gelelektrophorese unter reduzierenden Bedingungen (2.2.31): Die unter „Prüfung auf Identität“ beschriebenen Zonen müssen vorhanden sein.
Wasser (2.5.12): höchstens 5,0 Prozent, bestimmt mit 50 mg Substanz
Gehaltsbestimmung
Proteingehalt (2.5.33, Methode 4): 100 bis 700 mg · g–1
Untersuchungslösung: 10 mg Lyophilisat werden mit destilliertem Wasser R zu 1 ml verdünnt. Aus dieser Lösung werden weitere Verdünnungen hergestellt (zum Beispiel von 1 mg · ml–1 bis 5 mg · ml–1), sodass die Absorption bei der Messung innerhalb der Kalibrierkurve liegt.
Referenzlösungen: Rinderalbumin R 1 wird mit destilliertem Wasser R zu Konzentrationen im Bereich von 20 μg · ml–1 bis 2 000 μg · ml–1 verdünnt, zum Beispiel 25 μg · ml–1, 125 μg · ml–1, 250 μg · ml–1, 500 μg · ml–1, 750 μg · ml–1, 1 000 μg · ml–1, 1 500 μg · ml–1 und 2 000 μg · ml–1. Es müssen mindestens fünf Referenzlösungen hergestellt werden.
Die Gehaltsbestimmung kann alternativ auch mit einem käuflich erwerbbaren Kit durchgeführt werden.
Lagerung
Dicht verschlossen, bei Raumtemperatur
Arzneiformen
Herstellung
Lösung D1 und flüssige Verdünnungen nach Vorschrift 5a mit Gereinigtem Wasser oder nach Vorschrift 5b mit Wasser für Injektionszwecke
Verreibungen nach Vorschrift 6
Eigenschaften
Die Lösung D1 ist eine gelbbraune Flüssigkeit.
Die 1. Dezimalverreibung ist ein weißes bis hell gelbbraunes Pulver.
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (2.2.31): Die Bestimmung erfolgt wie unter „Prüfung auf Identität“ beschrieben mit 100 µl der 1. Dezimalverdünnung oder 100 mg der 1. Dezimalverreibung.
Prüfung auf Reinheit
Trockenrückstand (2.8.16): mindestens 9,0 Prozent für die Lösung D1
Equisetum hiemale
Equisetum hyemale
Verwendet werden die im Sommer gesammelten frischen ganzen Pflanzen von Equisetum hiemale L.
Beschreibung
Der aufrechte, sehr raue, unverzweigte, bis 1,50 m hohe Spross ist meist dunkelgrün, seltener etwas graugrün und hat einen Durchmesser von 4 bis 10 mm. Er setzt sich aus 3 bis 9 cm langen, flach gerippten Stängelgliedern zusammen, an deren Knoten Blätter sitzen, die zu einer stängelumfassenden, enganliegenden, walzenförmigen Scheide zusammengewachsen sind. Am Grunde und am Saum trägt sie eine schwarze Querbänderung. Die Spitzen der Blätter sind in jungem Stadium zu zugespitzten, weiß berandeten Zähnen ausgezogen, fallen jedoch schon frühzeitig ab, so dass nur noch ein stumpfer, gekerbter Rand sichtbar ist. Der spitz-kegelförmige, dunkelgraue Sporangienstand sitzt auf dem obersten, sehr kurzen Stängelglied und wird am Grunde von der obersten, glockenförmigen Scheide umschlossen.
Die unterirdischen Sprossteile entsprechen in ihrem morphologischen Aufbau den oberirdischen Sprossen, sind jedoch von schwarzbrauner Farbe und haben eine etwas kleinere Zentralhöhle. Die nur undeutlich rinnig erscheinenden, 2 bis 5 mm dicken Sprossachsen tragen an den Knoten 2 bis 4 mm hohe enganliegende, schwarzbraune Blattscheiden, an deren Basis gebüschelt feine, faserförmige, verzweigte Wurzeln entspringen. An der gleichen Stelle entspringen im unteren Bereich gelegentlich, in Nähe des Übergangs zu den oberirdischen Sprossachsen gehäuft, Seitenachsen. Die Internodien sind zum Teil in diesem Bereich sehr kurz, unregelmäßig knotig und mit Knospen neuer Seitenachsen besetzt.
Arzneiformen
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 2a
Eigenschaften
Die Urtinktur ist eine gelbbraun bis grün-braune Flüssigkeit.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: die Urtinktur
Referenzlösung: 2,5 mg Rutosid-Trihydrat R, 2,5 mg Hyperosid R und 1,0 mg Kaffeesäure R werden in 20 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R (5 bis 40 μm) [oder DC-Platte mit Kieselgel R (2 bis 10 μm)]
Fließmittel: wasserfreie Ameisensäure R, Essigsäure 99 % R, Wasser R, Ethylacetat R (7,5:7,5:18:67 V/V/V/V)
Auftragen: 20 μl [oder 5 μl] Untersuchungslösung und 5 μl [oder 5 μl] Referenzlösung; bandförmig 20 mm [oder 8 mm]
Laufstrecke: 10 cm [oder 6 cm]
Trocknen: 5 min lang im Kaltluftstrom
Detektion: Die Platte wird 3 min lang bei 100 °C erhitzt und noch warm mit einer Lösung von Diphenylboryloxylethylamin R (10 g · l–1) in Methanol R und anschließend mit einer Lösung von Macrogol 400 R (50 g · l–1) in Methanol R behandelt. Die Platte wird im Kaltluftstrom getrocknet. Die Auswertung erfolgt nach 10 min im ultravioletten Licht bei 365 nm.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können fluoreszierende weitere Zonen vorhanden sein.
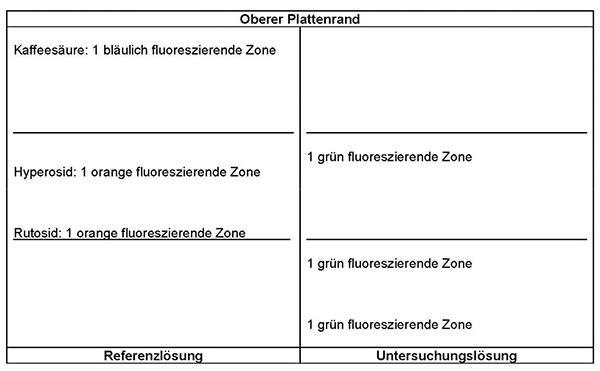
Prüfung auf Reinheit:
Equisetum arvense:
Im Chromatogramm der Untersuchungslösung (siehe Prüfung auf Identität) darf im mittleren Drittel keine orange fluoreszierende Zone vorhanden sein.
Relative Dichte (2.2.5): 0,930 bis 0,950
Trockenrückstand (H 2.2.6): mindestens 1,6 Prozent
Lagerung
Vor Licht geschützt
Mucor racemosus e volumine cellulae (lyophil., steril.)
Verwendet wird ein Lyophilisat, welches nach der Fermentation von Mucor racemosus f. sphaerosporus (Hagem) Schipper 1970 aus der vom Kulturmedium befreiten Biomasse hergestellt wird, mit einem Gehalt von mindestens 100 mg bis höchstens 700 mg Gesamtprotein pro Gramm Lyophilisat.
Herstellung
Die Biomasse wird nach den Grundprinzipien der Monographie Fermentationsprodukte (Ph. Eur.) gewonnen. Als Kulturmedium wird eine Mischung von Natriumnitrat (3,0 g · l–1), Eisen(II)-sulfat-Heptahydrat (0,01 g · l–1), Calciumchlorid-Dihydrat (0,05 g · l–1), Ammoniumsulfat (2,0 g · l–1), Kaliumchlorid (0,5 g · l–1), Kaliummonohydrogenphosphat (1,0 g · l–1), Maltose-Monohydrat (40,0 g · l–1), Caseinpepton (1,0 g · l–1), Hefeextrakt (1,0 g · l–1) und Magnesiumsulfat-Heptahydrat (0,5 g · l–1) in Gereinigtem Wasser verwendet. Die Fermentation erfolgt unter Zufuhr von Luft bei 24 °C bis 28 °C und einem pH-Wert von 5,0 bis 7,0. Die Ernte erfolgt am Ende der stammspezifischen Wachstumsphase zu Beginn der Abflachung der Turbidimetrie-Kurve. Die Biomasse wird durch Zentrifugation von dem Kulturmedium getrennt. Danach wird im gleichen Verfahren die Biomasse mit Gereinigtem Wasser versetzt, gewaschen und ebenfalls durch Zentrifugation abgetrennt. Das Waschvolumen muss mindestens dem 10-fachen des Produktvolumens nach Zellernte entsprechen. Anschließend werden die Zellen in einer geeigneten Zellmühle mechanisch aufgeschlossen. Diese Phase wird von den wasserunlöslichen Bestandteilen zunächst durch Zentrifugation und anschließend durch eine Mikrofiltration (0,2 μm) im Querstrom-Verfahren getrennt. Das resultierende Filtrat wird anschließend einer Sterilfiltration (0,2 μm) im Durchlauf-Verfahren unterzogen und muss der Prüfung auf Sterilität (2.6.1) entsprechen. Anschließend wird die Lösung gefriergetrocknet. Dieses Lyophilisat besteht aus wasserlöslichen Zellbestandteilen und wasserunlöslichen Zellbestandteilen, die kleiner als 0,2 μm sind.
Beschreibung
Makroskopische Merkmale: Die vegetative Form des Pilzes zeigt weiße Hyphen, mit dem Alter entstehen bräunlich graue Sporenträger.
Mikroskopische Merkmale: Die Sporenträger stehen auf den Hyphen, die häufig verzweigt sind. Die Sporen sind zuerst farblos und werden mit der Zeit braun. Die asexuellen Sporen sind im Mycel sowie in den Sporenträgern vorhanden.
Lyophilisat: Gelbbraunes, amorphes Pulver
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (2.2.31): unter Verwendung eines 4- bis 20-prozentigen Gradientengels
Die SDS-PAGE kann unter Verwendung von kommerziell erhältlichen Gradientengelen durchgeführt werden.
Gele mit Gradienten (Trenngele) enthalten eine von oben nach unten zunehmende Konzentration an Acrylamid. Die Herstellung von Gradientengelen erfolgt mit einer gradientenbildenden Apparatur.
Ein 4 bis 20 Prozent Acrylamid enthaltenes Gradientengel wird hergestellt.
Eine 4-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
10 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
72 Volumteilen Wasser R
- –
-
2 Volumteilen einer Lösung von Natriumdodecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Eine 20-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
50 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
15 Volumteilen Wasser R
- –
-
17 Volumteilen einer Lösung von Saccharose R (855,8 g · l–1)
- –
-
2 Volumteilen einer Lösung von Natriumdodecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Die Kammern der gradientenbildenden Apparatur werden mit den Acrylamid-Lösungen beladen. Das polymerisierte Gradientengel wird gemäß den Geräteangaben hergestellt.
Nach vollständiger Polymerisation wird das Gradientengel mit Wasser R gespült. Überschüssige Flüssigkeit wird entfernt. Die Lösung, die die Grundlage für das Anreicherungsgel bildet, wird in die Apparatur eingefüllt und ein sauberer Kamm wird eingelegt. Anschließend lässt man die Lösung des Anreicherungsgels polymerisieren.
Geldimension: 8,6 x 6,7 x 0,1 cm (Breite x Länge x Dicke)
SDS-PAGE-Laufpuffer (10x): 144 g Glycin R, 30 g Tris-Hydrochlorid (= Tris(hydroxymethyl)-aminomethan- bzw. Trometamolhydrochlorid) und 10 g Natriumdodecylsulfat R werden in Wasser R zu 1 000 ml gelöst. Der pH-Wert der Lösung sollte zwischen 8,1 und 8,8 liegen.
SDS-PAGE-Laufpuffer (1x): Vor Gebrauch wird der SDS-PAGE-Laufpuffer (10x) 1 zu 10 mit Wasser R verdünnt.
Tris-Hydrochlorid-Lösung (0,5 mol · l–1): 7,88 g Tris-Hydrochlorid wird mit Wasser R zu 100 ml verdünnt.
Natriumdodecylsulfat-Lösung (10 %): 5 g Natriumdodecylsulfat R werden mit Wasser R zu 50 ml verdünnt.
Bromphenolblau-Lösung (1 %): 0,05 g Bromphenolblau R werden mit Wasser R zu 5 ml verdünnt.
Proben-Pufferlösung: 20 ml Tris-Hydrochlorid–Lösung (0,5 mol · l–1), 16 ml Glycerol 85 % R 1, 32 ml Natriumdodecylsulfat-Lösung (10 %), 8 ml β-Mercaptoethanol R, 4 ml Bromphenolblau-Lösung (1 %) und 40 ml Wasser R werden gemischt.
Stammlösungen: Aus dem Lyophilisat wird eine Lösung hergestellt, die ungefähr 10 mg · ml–1 des Ausgangsmaterials enthält. Es können zusätzliche Verdünnungen wie 1 zu 2 oder 1 zu 5 hergestellt werden.
Untersuchungslösung: 150 µl der entsprechenden Stammlösung werden mit 50 µl Proben-Pufferlösung gemischt (beispielsweise in einem 1,5-ml-Eppendorf-Gefäß). Das Reaktionsgefäß wird verschlossen, in einem Wasserbad 5 min lang bei 95 °C erhitzt und danach auf Raumtemperatur abgekühlt.
Referenzlösung: Eine Lösung geeigneter Marker für Molekülmassen zur Kalibrierung von SDS-Polyacrylamidgelen im Bereich von 10 bis 250 kDa wird verwendet.
Detektion: Silberfärbung
Fixierlösung: 60 ml wasserfreies Ethanol R werden mit 20 ml wasserfreier Essigsäure R versetzt. Die Mischung wird mit 120 ml Wasser R verdünnt.
Silber-Färbelösung: 2 g Silbernitrat R und 0,2 ml Formaldehyd-Lösung R werden mit Wasser R zu 1 000 ml verdünnt.
Reduktionslösung: 314 mg Natriumthiosulfat R werden mit Wasser R zu 1 000 ml verdünnt.
Entwicklerlösung: 30 g wasserfreies Natriumcarbonat R, 0,5 ml Formaldehyd-Lösung R und 0,1 ml Reduktionslösung werden in Wasser R zu 1 000 ml verdünnt.
Blockierlösung: 5 g Glycin R werden in Wasser R zu 1 000 ml verdünnt.
Die Kassette mit dem Gel wird in die Elektrophorese-Apparatur eingebracht. Danach werden 600 ml des SDS-PAGE-Laufpuffers in die inneren und äußeren Kammern gefüllt. Dabei ist darauf zu achten, dass keine Luftblasen entstehen. Wenn notwendig, muss Puffer nachgefüllt werden. Danach wird der Kamm aus den Geltaschen entfernt und die Taschen mit SDS-PAGE-Laufpuffer gespült, um Luftbläschen oder nicht polymerisierte Acrylamid-Rückstände zu entfernen.
Auftragen: 10 µl Untersuchungslösung und 10 µl Referenzlösung
Laufbedingungen: Die Elektrophorese wird etwa 60 min lang bei Raumtemperatur unter Anlegen von 200 V durchgeführt. Diese Bedingungen gelten bei Verwendung eines Gels im oben genannten Format. Die Elektrophorese wird beendet, sobald die Farbstofffront die Referenzlinie auf der Kassette erreicht.
Der Lauf wird gestartet.
Silberfärbung: Nach dem Lauf wird das Gel aus der Kammer entfernt. Gegebenenfalls wird die Anleitung des Herstellers beachtet.
Folgende Schritte werden ausgeführt, wobei nach den Behandlungen die Lösungen jeweils vorsichtig abdekantiert werden:
- –
-
Fixieren: Das Gel wird 60 min lang unter leichtem Schütteln in Fixierlösung getaucht.
- –
-
Waschen: Das Gel wird 2-mal 20 min lang unter leichtem Schütteln in einer Mischung von 70 ml Wasser R und 30 ml wasserfreiem Ethanol R gewaschen.
- –
-
Waschen: Das Gel wird einmal 20 min lang unter leichtem Schütteln in Wasser R gewaschen.
- –
-
Reduktion: Das Gel wird etwa 1 min lang unter leichtem Schütteln in 100 ml Reduktionslösung getaucht.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Färben: Das Gel wird maximal 20 min lang unter leichtem Schütteln in Silber-Färbelösung getaucht. Das Gel bleibt nur so lange in der Färbelösung, bis sich eindeutige Banden ergeben. Danach kann der Färbeschritt beendet werden, auch wenn 20 min noch nicht abgelaufen sind.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Entwicklung: Das Gel wird etwa 3 bis 5 min lang unter leichtem Schütteln in 100 ml Entwicklerlösung getaucht.
- –
-
Waschen: Das Gel wird etwa 20 s lang unter leichtem Schütteln mit 100 ml Wasser R gewaschen.
- –
-
Blockieren: Das Gel wird etwa 10 min lang in Blockierlösung getaucht.
Ergebnis: Der Bereich der Molekülmassen und die Zonenfolge im Elektropherogramm der Untersuchungslösung sind aus den nachstehenden Angaben ersichtlich. Weitere Zonen können vorhanden sein.
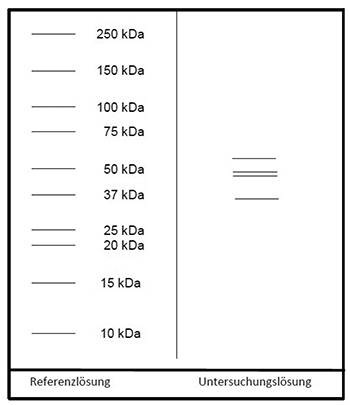
Prüfung auf Reinheit
Polyacrylamid-Gelelektrophorese unter reduzierenden Bedingungen (2.2.31): Die unter „Prüfung auf Identität“ beschriebenen Zonen müssen vorhanden sein.
Wasser (2.5.12): höchstens 5,0 Prozent, bestimmt mit 50 mg Substanz
Gehaltsbestimmung
Proteingehalt (2.5.33, Methode 4): 100 bis 700 mg · g–1
Untersuchungslösung: 10 mg Lyophilisat werden mit destilliertem Wasser R zu 1 ml verdünnt. Aus dieser Lösung werden weitere Verdünnungen hergestellt (zum Beispiel von 1 mg · ml–1 bis 5 mg ∙ ml–1), sodass die Absorption bei der Messung innerhalb der Kalibrierkurve liegt.
Referenzlösungen: Rinderalbumin R 1 wird mit destilliertem Wasser R zu Konzentrationen im Bereich von 20 μg · ml–1 bis 2 000 μg · ml–1 verdünnt, zum Beispiel 25 μg · ml–1, 125 μg · ml–1, 250 μg · ml–1, 500 μg · ml–1, 750 μg · ml–1, 1 000 μg · ml–1, 1 500 μg · ml–1und 2 000 μg · ml–1. Es müssen mindestens fünf Referenzlösungen hergestellt werden.
Die Gehaltsbestimmung kann alternativ auch mit einem käuflich erwerbbaren Kit durchgeführt werden.
Lagerung
Dicht verschlossen, bei Raumtemperatur
Arzneiformen
Herstellung
Lösung D1 und flüssige Verdünnungen nach Vorschrift 5a mit Gereinigtem Wasser oder nach Vorschrift 5b mit Wasser für Injektionszwecke
Verreibungen nach Vorschrift 6
Eigenschaften
Die Lösung D1 ist eine gelbbraune Flüssigkeit.
Die 1. Dezimalverreibung ist ein weißes bis hell gelbbraunes Pulver.
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (2.2.31): Die Bestimmung erfolgt wie unter „Prüfung auf Identität“ beschrieben mit 100 µl der 1. Dezimalverdünnung oder 100 mg der 1. Dezimalverreibung.
Prüfung auf Reinheit
Trockenrückstand (2.8.16): mindestens 9,0 Prozent für die Lösung D1
Penicillium chrysogenum e volumine cellulae (lyophil., steril.)
Verwendet wird ein Lyophilisat, welches nach der Fermentation von Penicillium chrysogenum Thom (1910) aus der vom Kulturmedium befreiten Biomasse hergestellt wird, mit einem Gehalt von mindestens 100 mg bis höchstens 600 mg Gesamtprotein pro Gramm Lyophilisat.
Herstellung
Die Biomasse wird nach den Grundprinzipien der Monographie Fermentationsprodukte (Ph. Eur.) gewonnen. Als Kulturmedium wird eine Mischung von Natriumnitrat (3,0 g · l–1), Eisen(II)-sulfat-Heptahydrat (0,01 g · l–1), Calciumchlorid-Dihydrat (0,05 g · l–1), Ammoniumsulfat (2,0 g · l–1), Kaliumchlorid (0,5 g · l–1), Kaliummonohydrogenphosphat (1,0 g · l–1), Maltose-Monohydrat (40,0 g · l–1), Caseinpepton (1,0 g · l–1), Hefeextrakt (1,0 g · l–1) und Magnesiumsulfat-Heptahydrat (0,5 g · l–1) in Gereinigtem Wasser verwendet. Die Fermentation erfolgt unter Zufuhr von Luft bei 24 °C bis 28 °C und einem pH-Wert von 4,5 bis 6,5. Die Ernte erfolgt am Ende der stammspezifischen Wachstumsphase zu Beginn der Abflachung der Turbidimetrie-Kurve. Die Biomasse wird durch Zentrifugation von dem Kulturmedium getrennt. Danach wird im gleichen Verfahren die Biomasse mit Gereinigtem Wasser versetzt, gewaschen und ebenfalls durch Zentrifugation abgetrennt. Das Waschvolumen muss mindestens dem 10-fachen des Produktvolumens nach Zellernte entsprechen. Anschließend werden die Zellen in einer geeigneten Zellmühle mechanisch aufgeschlossen. Diese Phase wird von den wasserunlöslichen Bestandteilen zunächst durch Zentrifugation und anschließend durch eine Mikrofiltration (0,2 μm) im Querstrom-Verfahren getrennt. Das resultierende Filtrat wird anschließend einer Sterilfiltration (0,2 μm) im Durchlauf-Verfahren unterzogen und muss der Prüfung auf Sterilität (2.6.1) entsprechen. Anschließend wird die Lösung gefriergetrocknet. Dieses Lyophilisat besteht aus wasserlöslichen Zellbestandteilen und wasserunlöslichen Zellbestandteilen, die kleiner als 0,2 μm sind.
Beschreibung
Makroskopische Merkmale: Die vegetative Form des Pilzes zeigt samtige Kolonieoberflächen, gelblich weiße Hyphen. Die Sporenträger verfärben sich mit der Zeit ins Gelbgrünliche bis Blaugrüne. Die Rückseite der Kolonie ist stark gelb bis blass cremefarben.
Mikroskopische Merkmale: Die Hyphen sind glatt, dünnwandig und durchsichtig.
Lyophilisat: Gelbbraunes, amorphes Pulver
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (2.2.31): unter Verwendung eines 4- bis 20-prozentigen Gradientengels
Die SDS-PAGE kann unter Verwendung von kommerziell erhältlichen Gradientengelen durchgeführt werden.
Gele mit Gradienten (Trenngele) enthalten eine von oben nach unten zunehmende Konzentration an Acrylamid. Die Herstellung von Gradientengelen erfolgt mit einer gradientenbildenden Apparatur.
Ein 4 bis 20 Prozent Acrylamid enthaltenes Gradientengel wird hergestellt.
Eine 4-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
10 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
72 Volumteilen Wasser R
- –
-
2 Volumteilen einer Lösung von Natriumdodecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Eine 20-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
50 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
15 Volumteilen Wasser R
- –
-
17 Volumteilen einer Lösung von Saccharose R (855,8 g · l–1)
- –
-
2 Volumteilen einer Lösung von Natriumdodecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Die Kammern der gradientenbildenden Apparatur werden mit den Acrylamid-Lösungen beladen. Das polymerisierte Gradientengel wird gemäß den Geräteangaben hergestellt.
Nach vollständiger Polymerisation wird das Gradientengel mit Wasser R gespült. Überschüssige Flüssigkeit wird entfernt. Die Lösung, die die Grundlage für das Anreicherungsgel bildet, wird in die Apparatur eingefüllt und ein sauberer Kamm wird eingelegt. Anschließend lässt man die Lösung des Anreicherungsgels polymerisieren.
Geldimension: 8,6 x 6,7 x 0,1 cm (Breite x Länge x Dicke)
SDS-PAGE-Laufpuffer (10x): 144 g Glycin R, 30 g Tris-Hydrochlorid (=Tris(hydroxymethyl)-aminomethan- bzw. Trometamolhydrochlorid) und 10 g Natriumdodecylsulfat R werden in Wasser R zu 1 000 ml gelöst. Der pH-Wert der Lösung sollte zwischen 8,1 und 8,8 liegen.
SDS-PAGE-Laufpuffer (1x): Vor Gebrauch wird der SDS-PAGE-Laufpuffer (10x) 1 zu 10 mit Wasser R verdünnt.
Tris-Hydrochlorid-Lösung (0,5 mol · l–1): 7,88 g Tris-Hydrochlorid wird mit Wasser R zu 100 ml verdünnt.
Natriumdodecylsulfat-Lösung (10 %): 5 g Natriumdodecylsulfat R werden mit Wasser R zu 50 ml verdünnt.
Bromphenolblau-Lösung (1 %): 0,05 g Bromphenolblau R werden mit Wasser R zu 5 ml verdünnt.
Proben-Pufferlösung: 20 ml Tris-Hydrochlorid-Lösung (0,5 mol · l–1), 16 ml Glycerol 85 % R 1, 32 ml Natriumdodecylsulfat-Lösung (10 %), 8 ml β-Mercaptoethanol R, 4 ml Bromphenolblau-Lösung (1 %) und 40 ml Wasser R werden gemischt.
Stammlösungen: Aus dem Lyophilisat wird eine Lösung hergestellt, die ungefähr 10 mg · ml–1 des Ausgangsmaterials enthält. Es können zusätzliche Verdünnungen wie 1 zu 2 oder 1 zu 5 hergestellt werden.
Untersuchungslösung: 150 µl der entsprechenden Stammlösung werden mit 50 µl Proben-Pufferlösung gemischt (beispielsweise in einem 1,5-ml-Eppendorf-Gefäß). Das Reaktionsgefäß wird verschlossen, in einem Wasserbad 5 min lang bei 95 °C erhitzt und danach auf Raumtemperatur abgekühlt.
Referenzlösung: Eine Lösung geeigneter Marker für Molekülmassen zur Kalibrierung von SDS-Polyacrylamidgelen im Bereich von 10 bis 250 kDa wird verwendet.
Detektion: Silberfärbung
Fixierlösung: 60 ml wasserfreies Ethanol R werden mit 20 ml wasserfreier Essigsäure R versetzt. Die Mischung wird mit 120 ml Wasser R verdünnt.
Silber-Färbelösung: 2 g Silbernitrat R und 0,2 ml Formaldehyd-Lösung R werden mit Wasser R zu 1 000 ml verdünnt.
Reduktionslösung: 314 mg Natriumthiosulfat R werden mit Wasser R zu 1 000 ml verdünnt.
Entwicklerlösung: 30 g wasserfreies Natriumcarbonat R, 0,5 ml Formaldehyd-Lösung R und 0,1 ml Reduktionslösung werden in Wasser R zu 1 000 ml verdünnt.
Blockierlösung: 5 g Glycin R werden in Wasser R zu 1 000 ml verdünnt.
Die Kassette mit dem Gel wird in die Elektrophorese-Apparatur eingebracht. Danach werden 600 ml des SDS-PAGE-Laufpuffers in die inneren und äußeren Kammern gefüllt. Dabei ist darauf zu achten, dass keine Luftblasen entstehen. Wenn notwendig, muss Puffer nachgefüllt werden. Danach wird der Kamm aus den Geltaschen entfernt und die Taschen mit SDS-PAGE-Laufpuffer gespült, um Luftbläschen oder nicht polymerisierte Acrylamid-Rückstände zu entfernen.
Auftragen: 10 µl Untersuchungslösung und 10 µl Referenzlösung
Laufbedingungen: Die Elektrophorese wird etwa 60 min lang bei Raumtemperatur unter Anlegen von 200 V durchgeführt. Diese Bedingungen gelten bei Verwendung eines Gels im oben genannten Format. Die Elektrophorese wird beendet, sobald die Farbstofffront die Referenzlinie auf der Kassette erreicht.
Der Lauf wird gestartet.
Silberfärbung: Nach dem Lauf wird das Gel aus der Kammer entfernt. Gegebenenfalls wird die Anleitung des Herstellers beachtet.
Folgende Schritte werden ausgeführt, wobei nach den Behandlungen die Lösungen jeweils vorsichtig abdekantiert werden:
- –
-
Fixieren: Das Gel wird 60 min lang unter leichtem Schütteln in Fixierlösung getaucht.
- –
-
Waschen: Das Gel wird 2-mal 20 min lang unter leichtem Schütteln in einer Mischung von 70 ml Wasser R und 30 ml wasserfreiem Ethanol R gewaschen.
- –
-
Waschen: Das Gel wird einmal 20 min lang unter leichtem Schütteln in Wasser R gewaschen.
- –
-
Reduktion: Das Gel wird etwa 1 min lang unter leichtem Schütteln in 100 ml Reduktionslösung getaucht.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Färben: Das Gel wird maximal 20 min lang unter leichtem Schütteln in Silber-Färbelösung getaucht. Das Gel bleibt nur so lange in der Färbelösung, bis sich eindeutige Banden ergeben. Danach kann der Färbeschritt beendet werden, auch wenn 20 min noch nicht abgelaufen sind.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Entwicklung: Das Gel wird etwa 3 bis 5 min lang unter leichtem Schütteln in 100 ml Entwicklerlösung getaucht.
- –
-
Waschen: Das Gel wird etwa 20 s lang unter leichtem Schütteln mit 100 ml Wasser R gewaschen.
- –
-
Blockieren: Das Gel wird etwa 10 min lang in Blockierlösung getaucht.
Ergebnis: Der Bereich der Molekülmassen und die Zonenfolge im Elektropherogramm der Untersuchungslösung sind aus den nachstehenden Angaben ersichtlich. Weitere Zonen können vorhanden sein.
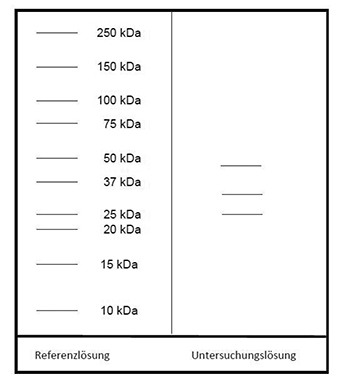
Prüfung auf Reinheit
Polyacrylamid-Gelelektrophorese unter reduzierenden Bedingungen (2.2.31): Die unter „Prüfung auf Identität“ beschriebenen Zonen müssen vorhanden sein.
Wasser (2.5.12): höchstens 5,0 Prozent, bestimmt mit 50 mg Substanz
Gehaltsbestimmung
Proteingehalt (2.5.33, Methode 4): 100 bis 600 mg · g–1
Untersuchungslösung: 10 mg Lyophilisat werden mit destilliertem Wasser R zu 1 ml verdünnt. Aus dieser Lösung werden weitere Verdünnungen hergestellt (zum Beispiel von 1 mg · ml–1 bis 5 mg · ml–1), sodass die Absorption bei der Messung innerhalb der Kalibrierkurve liegt.
Referenzlösungen: Rinderalbumin R 1 wird mit destilliertem Wasser R zu Konzentrationen im Bereich von 20 μg · ml–1 bis 2 000 μg · ml–1 verdünnt, zum Beispiel 25 μg · ml–1, 125 μg · ml–1, 250 μg · ml–1, 500 μg · ml–1, 750 μg · ml–1, 1 000 μg · ml–1, 1 500 μg · ml–1 und 2 000 μg · ml–1. Es müssen mindestens fünf Referenzlösungen hergestellt werden.
Die Gehaltsbestimmung kann alternativ auch mit einem käuflich erwerbbaren Kit durchgeführt werden.
Lagerung
Dicht verschlossen, bei Raumtemperatur
Arzneiformen
Herstellung
Lösung D1 und flüssige Verdünnungen nach Vorschrift 5a mit Gereinigtem Wasser oder nach Vorschrift 5b mit Wasser für Injektionszwecke
Verreibungen nach Vorschrift 6
Eigenschaften
Die Lösung D1 ist eine gelbbraune Flüssigkeit.
Die 1. Dezimalverreibung ist ein weißes bis hell gelbbraunes Pulver.
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (2.2.31): Die Bestimmung erfolgt wie unter „Prüfung auf Identität“ beschrieben mit 100 µl der 1. Dezimalverdünnung oder 100 mg der 1. Dezimalverreibung.
Prüfung auf Reinheit
Trockenrückstand (2.8.16): mindestens 9,0 Prozent für die Lösung D1
Revidierte Monographien
Anmerkung zur Monographie:
Die relative Dichte wird aufgrund der vorgelegten Chargendaten und in Analogie zu der Spezifikation der DAC-Monographie „Ameisensäure 25 Prozent“ auf 1,058 bis 1,063 geändert.
Acidum formicicum
CH2O2
M r 46,03
Verwendet wird Ameisensäure, die mindestens 24,0 und höchstens 25,0 Prozent (m/m) CH2O2 enthält.
Eigenschaften
Klare, farblose, flüchtige Flüssigkeit mit stechendem Geruch und einer in starker Verdünnung noch saurer Reaktion; mischbar mit Wasser und Ethanol 96 % (V/V), nicht mischbar mit Ether
Prüfung auf Identität
- A.
-
2,0 ml verdünnte Natriumhydroxid-Lösung R werden mit 0,05 ml Phenolphtalein-Lösung R versetzt, gemischt und tropfenweise mit der Substanz versetzt, bis die Lösung farblos ist. 1 ml dieser Lösung wird mit 0,5 ml Silbernitrat-Lösung R 2 versetzt und gemischt. Zunächst tritt keine Änderung auf. Langsam (nach Erhitzen schneller) wird die Lösung grau und trübe durch die Ausfällung metallischen Silbers.
- B.
-
Die unter „Prüfung auf Identität, A“ verbliebene neutralisierte Lösung wird mit 0,5 ml Eisen(III)‐chlorid‐Lösung R 1 versetzt. Die Lösung färbt sich orangerot bis bräunlich rot.
Prüfung auf Reinheit
Prüflösung: 10,0 ml Substanz werden mit 25,0 ml verdünnter Natriumhydroxid-Lösung R versetzt. Die Mischung wird mit destilliertem Wasser R zu 50,0 ml verdünnt.
Aussehen der Substanz: Die Substanz muss klar (2.2.1) und farblos (2.2.2, Methode II) sein.
Verwandte Substanzen: Flüssigchromatographie (2.2.29)
Untersuchungslösung: 2,000 g Substanz werden mit der mobilen Phase zu 100,0 ml verdünnt.
Referenzlösung a: 0,500 g wasserfreie Essigsäure R (Verunreinigung A) werden mit der mobilen Phase zu 100,0 ml verdünnt. 2,0 ml Lösung und 1,0 ml Untersuchungslösung werden gemischt und mit der mobilen Phase zu 100,0 ml verdünnt.
Referenzlösung b: 1,0 ml Untersuchungslösung wird mit der mobilen Phase zu 100,0 ml verdünnt. 1,0 ml dieser Lösung wird mit der mobilen Phase zu 10,0 ml verdünnt.
Säule
- –
-
Größe: l = 0,25 m, Ø = 4,6 mm
- –
-
Stationäre Phase: nachsilanisiertes, octadecylsilyliertes, mit zu 100 Prozent wässrigen mobilen Phasen kompatibles Kieselgel zur Chromatographie R (4 µm)
Mobile Phase: Eine Lösung von Kaliumdihydrogenphosphat R (2,72 g · l–1) wird mit Phosphorsäure 10 % R auf einen pH-Wert von 2,9 eingestellt.
Durchflussrate: 1,0 ml · min–1
Detektion: Spektrometer bei 220 nm
Einspritzen: 20 µl
Chromatographiedauer: 3fache Retentionszeit von Ameisensäure
Relative Retention (bezogen auf Ameisensäure, tR etwa 3 min)
- –
-
Verunreinigung A: etwa 1,5
Eignungsprüfung: Referenzlösung a
- –
-
Auflösung: mindestens 5,0 zwischen den Peaks von Ameisensäure und Verunreinigung A
Grenzwerte
- –
-
Nicht spezifizierte Verunreinigungen: jeweils nicht größer als die Fläche des Hauptpeaks im Chromatogramm der Referenzlösung b (0,10 Prozent)
- –
-
Summe aller Verunreinigungen: nicht größer als das 3fache der Fläche des Hauptpeaks im Chromatogramm der Referenzlösung b (0,3 Prozent)
- –
-
Ohne Berücksichtigung bleiben: Peaks, deren Fläche nicht größer ist als das 0,5fache der Fläche des Hauptpeaks im Chromatogramm der Referenzlösung b (0,05 Prozent).
Relative Dichte (2.2.5): 1,058 bis 1,063
Chlorid (2.4.4): 2,50 g Substanz werden mit Wasser R zu 15,0 ml verdünnt. Die Lösung muss der Grenzprüfung auf Chlorid entsprechen (20 ppm).
Sulfat (2.4.13): 15 ml Prüflösung müssen der Grenzprüfung auf Sulfat entsprechen (50 ppm).
Sulfit: 10 ml Prüflösung dürfen 0,5 ml Iod-Lösung (0,005 mol · l−1) RH nicht entfärben.
Schwermetalle (2.4.8): 12 ml Prüflösung müssen der Grenzprüfung A auf Schwermetalle entsprechen (10 ppm). Zur Herstellung der Referenzlösung wird die Blei-Lösung (2 ppm Pb) R verwendet.
Nicht flüchtige Verunreinigungen: höchstens 5 mg je 100 ml
20,0 ml Substanz werden auf dem Wasserbad zur Trockne eingedampft. Der Rückstand wird bei 100 bis 105 °C bis zur Massekonstanz getrocknet.
Gehaltsbestimmung
5,000 g Substanz werden mit 20 ml Wasser R verdünnt und nach Zusatz von 1 ml Phenolphthalein-Lösung R mit Natriumhydroxid-Lösung (1 mol · l−1) titriert.
1 ml Natriumhydroxid-Lösung (1 mol · l−1) entspricht 46,03 mg CH2O2.
Arzneiformen
Die Lösung D1 enthält mindestens 9,1 und höchstens 10,5 Prozent CH2O2.
Herstellung
Lösung D1 nach Vorschrift 5a aus 40 Teilen Substanz und 60 Teilen Gereinigtem Wasser
Die 2. Dezimalverdünnung wird mit Gereinigtem Wasser, die folgenden Verdünnungen werden mit Ethanol 43 % (m/m) hergestellt.
Eigenschaften
Die Lösung D1 ist eine klare, farblose Flüssigkeit mit stechendem Geruch.
Prüfung auf Identität
Die Lösung D1 gibt die Identitätsreaktionen der Substanz.
Prüfung auf Reinheit
Aussehen der Lösung: Die Lösung D1 muss klar (2.2.1) und farblos (2.2.2, Methode II) sein.
Relative Dichte (2.2.5): 1,023 bis 1,027
Gehaltsbestimmung
Die Bestimmung erfolgt wie unter „Gehaltsbestimmung“ bei der Substanz angegeben mit 10,00 g der Lösung D1.
Lagerung
Lösung D1 in Glasschliffflaschen oder anderen geeigneten Behältnissen
Anmerkungen zur Monographie:
- –
-
Definition: Änderung dahingehend, dass neben geschälter Droge auch ungeschälte Droge verwendet werden kann. Hintergrund sind einerseits die in den letzten Jahren bestehenden Probleme mit der Beschaffung von geschälter Droge und andererseits, dass z. T. noch geschälte Droge eingesetzt wird. Bei Verwendung von geschälter und ungeschälter Droge ergeben sich keine Unterschiede in den Qualitätsparametern.In den Arzneibüchern von Österreich und der Schweiz wird ebenfalls einmal geschälte und einmal ungeschälte Droge beschrieben.
- –
-
Definition: Absenkung des Mindestgehalts an ätherischem Öl auf 15 ml/kg basierend auf den vorgelegten Chargendaten
- –
-
Überarbeitung der makros- und mikroskopischen Pflanzenbeschreibung für geschälte Droge, ungeschälte Droge und Schnittdroge
- –
-
Prüfung auf Identität:
- –
-
Streichung der nasschemischen Identitätsreaktion
- –
-
Ersatz der DC-Beschreibung durch eine schematische Darstellung (DC-Kasten) und Ergänzung der HPTLC-Bedingungen
Acorus calamus
Calamus aromaticus
Verwendet wird der, von den Wurzeln und Blattresten befreite, getrocknete Wurzelstock von Acorus calamus L. 1 kg Droge enthält mindestens 15 ml ätherisches Öl.
Beschreibung
Die Droge hat einen aromatischen Geruch.
Ungeschälter Wurzelstock:
Die Oberfläche des Wurzelstocks ist hell- bis dunkelbraun und längs gestreift. Im Querschnitt weisen die innen liegenden Gewebeteile eine gelblich weiße, hellbraune bis rötlich weiße Farbe auf. Der Wurzelstock ist hart und von mehr oder weniger zylindrischem Querschnitt.
An der morphologischen Unterseite sind die Wurzelansätze als kleine, rundliche bis ovale, scharfrandige, hell- bis dunkelbraune, in unregelmäßigen Zickzacklinien angeordnete Narben zu erkennen; an der Oberseite und den Flanken sind Blattnarben in Form spitz-dreieckiger oder streifenförmiger, brauner Felder vorhanden, in deren Achseln Sprossnarben sitzen. Auf dem Querschnitt hebt sich der dunklere, von vielen Leitbündeln punktiert erscheinende Zentralzylinder von der helleren, ebenfalls Leitbündel tragenden Rinde deutlich ab.
Geschälter Wurzelstock:
Die Oberfläche des geschälten Wurzelstocks ist gelblich weiß, hellbraun bis rötlich weiß. Der Wurzelstock ist hart und von mehr oder weniger zylindrischem Querschnitt.
An der morphologischen Unterseite sind undeutlich bis deutlich die Wurzelansätze als kleine, rundliche bis ovale, hell- bis dunkelbraune Narben in unregelmäßigen Zickzacklinien zu erkennen. Auf dem Querschnitt hebt sich der dunklere, von vielen Leitbündeln punktiert erscheinende Zentralzylinder von der helleren, ebenfalls Leitbündel tragenden Rinde deutlich ab.
Schnittdroge:
Ungeschälter Wurzelstock:
Die Schnittdroge ist charakterisiert durch gelblich weiße, hellbraun bis rötlich weiße, unregelmäßig geformte Stücke. Diese lassen stellenweise an der hell- bis dunkelbraunen und gelegentlich längs gestreiften Außenseite Blattnarben und/oder die in unregelmäßigen Zickzacklinien angeordneten, rundlichen bis ovalen, hell- bis dunkelbraunen Wurzelnarben erkennen. Der durch die Leitbündel punktiert erscheinende Zentralzylinder hebt sich durch seine abweichende Färbung von der Rinde, in der auch Leitbündel vorkommen können, ab.
Geschälter Wurzelstock:
Die Schnittdroge ist charakterisiert durch gelblich weiße, hellbraun bis rötlich weiße, unregelmäßig geformte Stücke. Diese zeigen vereinzelt an der Außenseite, in unregelmäßigen Zickzacklinien angeordnete, rundliche bis ovale, hell- bis dunkelbraune undeutlich bis deutlich erkennbare Wurzelnarben. Der durch die Leitbündel punktiert erscheinende Zentralzylinder hebt sich durch seine abweichende Färbung von der Rinde, in der auch Leitbündel vorkommen können, ab.
Mikroskopische Merkmale:
Der ungeschälte Wurzelstock weist eine kleinzellige, oft derbwandige Epidermis auf und ein Korkgewebe, das nur um die Blatt- und Wurzelnarben herum ausgebildet ist. Bei der geschälten Droge sind weder Epidermis noch Kork vorhanden. Das Grundgewebe des Wurzelstocks besteht überwiegend aus einem interzellularenreichen Parenchym aus rundlich-polygonalen Zellen, die in der Regel 1 bis 4 µm, selten bis 10 µm große, einzelne, gelegentlich zu zwei bis vier zusammengesetzte, rundliche bis unregelmäßig elliptische Stärkekörner enthalten. An den Ansatzstellen zusammengesetzter Stärkekörner können diese auch kantig ausgebildet sein. Durch die in Längsrichtung zylindrisch gestreckten Interzellularräume erscheint das Parenchym im Querschnitt netzartig. Vorzugsweise an den Kreuzungspunkten dieses Netzes liegen Exkretzellen mit ätherischem Öl, verkorkter Wand und stark lichtbrechendem Inhalt oder gelegentlich auch gerbstoffhaltige Exkretzellen mit klumpenförmigem, braunem Inhalt. Die außerhalb der einschichtigen, wenig verdickten Endodermis verlaufenden Leitbündel sind in der Regel kollateral, haben englumige Schrauben- und Tüpfelgefäße und sind von einem Mantel verdickter Fasern und zuweilen von Kristallzellreihen mit Einzelkristallen begleitet. Die in der Nähe der Endodermis gehäuft auftretenden Leitbündel des Zentralzylinders sind in der Regel konzentrisch mit Innenphloem und einem lockeren Ring von Gefäßen mit ring-, treppen- oder netzförmiger Wandverdickung und haben in der Regel keinen Faserbelag.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: 1 g pulverisierte Droge (710) wird mit 10 ml Ethanol 70 % R versetzt. Die Mischung wird auf dem Wasserbad zum Sieden erhitzt und nach dem Erkalten filtriert.
Referenzlösung: 30 mg Hydrochinon R, 10 mg Borneol R und 10 mg Anethol R werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R (5 bis 40 µm) [oder DC-Platte mit Kieselgel R (2 bis 10 μm)]
Fließmittel: Ethylacetat R, Hexan R (28:72 V/V)
Auftragen: 30 µl [oder 8 µl] Untersuchungslösung und 10 µl [oder 3 μl] Referenzlösung; bandförmig 20 mm [oder 10 mm]
Laufstrecke: 10 cm [oder 6 cm]
Detektion: Nach Verdunsten des Fließmittels wird die Platte mit Anisaldehyd-Reagenz R behandelt und 5 bis 10 min lang bei 100 bis 105 °C erhitzt. Die Auswertung erfolgt innerhalb von 10 min im Tageslicht.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
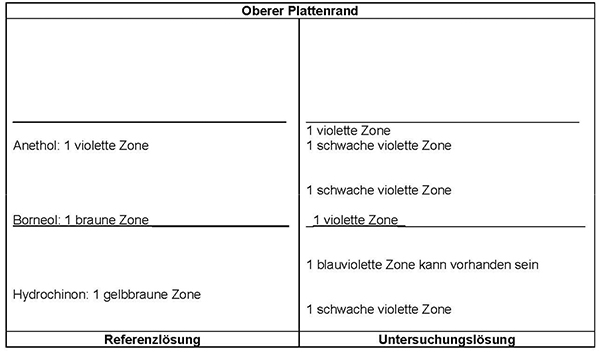
Prüfung auf Reinheit
cis-Isoasaron: höchstens 0,5 Prozent
1,00 g pulverisierte Droge (710) wird mit 40 ml Hexan R versetzt. Die Mischung wird 1 h lang gerührt und anschließend durch ein mittelhartes Filter in einen 50-ml-Messkolben filtriert. Das Filtrat wird unter Nachspülen des Filters mit Hexan R aufgefüllt. 5,0 ml Lösung werden in einem 25-ml-Messkolben mit Hexan R aufgefüllt. Die Absorption A (2.2.25) dieser Lösung wird bei 253 nm und bei 303 nm gegen Hexan R als Kompensationsflüssigkeit gemessen.
Folgende Forderungen müssen erfüllt werden:
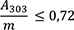
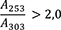
- A 253
-
= Absorption der Lösung bei 253 nm
- A 303
-
= Absorption der Lösung bei 303 nm
- m
-
= Einwaage der Droge in Gramm
Fremde Bestandteile (2.8.2): höchstens 2 Prozent
Asche (2.4.16): höchstens 6,0 Prozent
Gehaltsbestimmung
Ätherisches Öl (2.8.12): Die Bestimmung erfolgt mit 15,0 g unmittelbar zuvor pulverisierter Droge (710) und 200 ml Wasser R in einem 500-ml-Rundkolben und mit 0,5 ml Xylol R als Vorlage. Die Destillation erfolgt 4 h lang mit einer Destillationsgeschwindigkeit von 2 bis 3 ml je Minute.
Arzneiformen
Herstellung
Urtinktur aus der pulverisierten Droge (710) und flüssige Verdünnungen nach Vorschrift 4a mit Ethanol 62 % (m/m)
Eigenschaften
Die Urtinktur ist eine hellgelbe Flüssigkeit mit scharfem, charakteristischem Geruch.
Prüfung auf Identität
Die Urtinktur gibt die Identitätsreaktion der Droge. Untersuchungslösung ist die Urtinktur.
Prüfung auf Reinheit
cis-Isoasaron: höchstens 0,05 Prozent
4,00 g Urtinktur werden mit 15 ml Wasser R in einen Scheidetrichter überführt. Nach Zusatz von 1 g Natriumchlorid R wird die Mischung 4-mal mit je 20 ml Hexan R ausgeschüttelt. Die organischen Phasen werden in einem 100-ml-Messkolben vereinigt und mit Hexan R aufgefüllt. Die Absorption A (2.2.25) dieser Lösung wird bei 253 nm und bei 303 nm gegen Hexan R als Kompensationsflüssigkeit gemessen.
Folgende Forderungen müssen erfüllt werden:
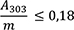
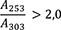
- A 253
-
= Absorption der Lösung bei 253 nm
- A 303
-
= Absorption der Lösung bei 303 nm
- m
-
= Einwaage der Urtinktur in Gramm
Relative Dichte (2.2.5): 0,885 bis 0,905
Trockenrückstand (H 2.2.6): mindestens 1,5 Prozent
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Bei der Reinheitsprüfung auf Cadmium und Quecksilber wird die Möglichkeit zur Prüfung an der Urtinktur aufgenommen, vergleichbar wie in der Ph.-Eur.-Monographie „Pflanzliche Drogen für homöopathische Zubereitungen“. Hintergrund hierfür ist, dass die Grenzwerte am Ausgangsmaterial häufig überschritten werden. Die Festlegung von höheren Grenzwerten ist jedoch schwierig, da die vorliegenden Schwermetalldaten eine relativ große Spanne aufweisen und nicht normalverteilt sind.
Ambra grisea
Ambra
Verwendet wird die vom Pottwal, Physeter catodon L. (Syn. Physeter macrocephalus L.) in den Eingeweiden abgesonderte und ausgeschiedene Substanz.
Beschreibung
Die Substanz hat einen eigentümlich aromatischen Geruch.
Sie besteht aus großen, rundlichen Brocken von graubrauner bis schwarzer Farbe. Die Oberfläche ist matt, klebrig, pechartig und fühlt sich fettig an. In der Wärme lässt sie sich kneten und schneiden, wird bei 60 °C salbenartig und verflüssigt sich in siedendem Wasser. Der Bruch ist im frischen Zustand honig- bis orangegelb. Der Schnitt weist konzentrische, verschiedenfarbige Schichtung auf. Die Substanz ist durchsetzt mit einzelnen dunkelbraunen, papageischnabelförmigen Hornkiefern. Im ultravioletten Licht bei 365 nm fluoresziert das Innere gelblich, ziegelrot oder orange.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: 1 g zerkleinerte Substanz wird mit 10 ml wasserfreiem Ethanol R versetzt, die Mischung 30 min lang gerührt und anschließend filtriert.
Referenzlösung: 10 mg Cholesterol R und 10 mg Bornylacetat R werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R
Fließmittel: Ether R, Toluol R (20:80 V/V)
Auftragen: 20 µl Untersuchungslösung und 10 µl Referenzlösung; bandförmig 20 mm
Laufstrecke: 10 cm
Detektion: Nach Verdunsten des Fließmittels wird die Platte mit Anisaldehyd-Reagenz R behandelt und 5 bis 10 min lang bei 100 bis 105 °C erhitzt. Die Auswertung erfolgt innerhalb von 10 min im Tageslicht.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
Prüfung auf Reinheit
Cadmium (2.4.27): höchstens 10 ppm
Quecksilber (2.4.27): höchstens 1 ppm
In begründeten Fällen kann die Prüfung auf Cadmium und Quecksilber mit der Urtinktur entsprechend den Anforderungen der allgemeinen Monographie Urtinkturen für homöopathische Zubereitungen durchgeführt werden. Dabei sind die oben genannten Grenzwerte für das Ausgangsmaterial unter Berücksichtigung des Verdünnungsgrades der Urtinktur und der Bestimmungsgrenze der Methode zugrunde zu legen.
Fremde Bestandteile (2.8.2): Pflanzenteile dürfen nicht vorhanden sein.
Asche (2.4.16): höchstens 3,0 Prozent
Arzneiformen
Herstellung
Zur Herstellung der Urtinktur D1 werden 10 Teile zerkleinerte Substanz mit 100 Teilen wasserfreiem Ethanol versetzt. Die Mischung wird eine Stunde lang unter Rückflusskühlung zum Sieden erhitzt und nach dem Erkalten filtriert.
Die 2. und 3. Dezimalverdünnung werden mit wasserfreiem Ethanol, die 4. Dezimalverdünnung wird mit Ethanol 86 % (m/m) und die folgenden Verdünnungen werden mit Ethanol 43 % (m/m) hergestellt.
Eigenschaften
Die Urtinktur ist eine goldbraune Flüssigkeit.
Prüfung auf Identität
Die Urtinktur gibt die Identitätsreaktion der Substanz. Untersuchungslösung ist die Urtinktur.
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,793 bis 0,813
Trockenrückstand (H 2.2.6): mindestens 7,5 Prozent
Lagerung
Dicht verschlossen, vor Licht geschützt
Anmerkungen zur Monographie:
In Analogie zu den drei neuen Monographien für Pilzlyophilisate wurden im Abschnitt „Herstellung“ folgende Formulierungen präzisiert:
- –
-
Nach dem mechanischen Aufschluss der Zellen ist die bisherige Formulierung „flüssige Phase“ auf „diese Phase“ geändert worden, da die Phase aus Wasser und Zellbestandteilen besteht.
- –
-
Es wird präzisiert, dass das Lyophilisat nicht nur aus wasserlöslichen Zellbestandteilen besteht, sondern auch wasserunlösliche Zellbestandteile enthält, die kleiner als 0,2 μm sind.
Aspergillus niger e volumine cellulae (lyophil., steril.)
Verwendet wird ein Lyophilisat, welches nach der Fermentation von Aspergillus niger van Tieghem aus der vom Kulturmedium befreiten Biomasse hergestellt wird, mit einem Gehalt von mindestens 100 mg bis höchstens 500 mg Gesamtprotein pro Gramm Lyophilisat.
Herstellung
Die Biomasse wird nach den Grundprinzipien der Monographie Fermentationsprodukte (Ph. Eur.) gewonnen. Als Kulturmedium wird eine Mischung von Natriumnitrat (3,0 g · l–1), Eisen(II)-sulfat-Heptahydrat (0,01 g · l–1), Calciumchlorid-Dihydrat (0,05 g · l–1), Ammoniumsulfat (2,0 g · l–1), Kaliumchlorid (0,5 g · l–1), Kaliummonohydrogenphosphat (1,0 g · l–1), Maltose-Monohydrat (50,0 g · l–1), Caseinpepton (1,0 g · l–1), Magnesiumsulfat-Heptahydrat (0,5 g · l–1) und Hefeextrakt (1,0 g · l–1) in Gereinigtem Wasser verwendet. Die Fermentation erfolgt unter Zufuhr von Luft bei 24 °C bis 28 °C und einem pH-Wert von 4,5 bis 6,5. Die Ernte erfolgt am Ende der stammspezifischen Wachstumsphase zu Beginn der Abflachung der Turbidimetrie-Kurve. Die Biomasse wird durch Zentrifugation von dem Kulturmedium getrennt. Danach wird im gleichen Verfahren die Biomasse mit Gereinigtem Wasser versetzt, gewaschen und ebenfalls durch Zentrifugation abgetrennt. Das Waschvolumen muss mindestens dem 10-fachen des Produktvolumens nach Zellernte entsprechen. Anschließend werden die Zellen in einer geeigneten Zellmühle mechanisch aufgeschlossen. Diese Phase wird von den wasserunlöslichen Bestandteilen zunächst durch Zentrifugation und anschließend durch eine Mikrofiltration (0,2 µm) im Querstrom-Verfahren getrennt. Das resultierende Filtrat wird anschließend einer Sterilfiltration (0,2 µm) im Durchlauf-Verfahren unterzogen und muss der Prüfung auf Sterilität (2.6.1) entsprechen. Anschließend wird die Lösung gefriergetrocknet. Dieses Lyophilisat besteht aus wasserlöslichen Zellbestandteilen und wasserunlöslichen Zellbestandteilen, die kleiner als 0,2 μm sind.
Beschreibung
Makroskopische Merkmale: Die vegetative Form des Pilzes zeigt filzähnliche, gelblich weiße Hyphen. Nach Bildung der Konidien nehmen die Hyphen eine typische schwarze Farbe an.
Mikroskopische Merkmale: Hyaline, septierte Hyphen, lange asexuelle Konidiophoren mit kugeliger Spitze.
Lyophilisat: weißes bis gelblich braunes amorphes Pulver
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (2.2.31): unter Verwendung eines 4- bis 20-prozentigen Gradientengels
Die SDS-PAGE kann unter Verwendung von kommerziell erhältlichen Gradientengelen durchgeführt werden.
Gele mit Gradienten (Trenngele) enthalten eine von oben nach unten zunehmende Konzentration an Acrylamid. Die Herstellung von Gradientengelen erfolgt mit einer gradientenbildenden Apparatur.
Ein 4 bis 20 Prozent Acrylamid enthaltenes Gradientengel wird hergestellt.
Eine 4-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
10 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
72 Volumteilen Wasser R
- –
-
2 Volumteilen einer Lösung von Natriumdodecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Eine 20-prozentige Acrylamid-Lösung wird hergestellt durch Mischen von
- –
-
50 Volumteilen einer 40-prozentigen Acrylamid-Bisacrylamid-Lösung (37,5:1 V/V)
- –
-
13 Volumteilen Trometamol-Pufferlösung pH 8,8 (3 mol · l–1) R
- –
-
15 Volumteilen Wasser R
- –
-
17 Volumteilen einer Lösung von Saccharose R (855,8 g · l–1)
- –
-
2 Volumteilen einer Lösung von Natriumdocecylsulfat R (100 g · l–1)
- –
-
1 Volumteil einer Lösung von Putrescin R (0,04 mg · l–1)
- –
-
2 Volumteilen einer Lösung von Ammoniumpersulfat R (15 mg · l–1)
Die Kammern der gradientenbildenden Apparatur werden mit den Acrylamid-Lösungen beladen. Das polymerisierte Gradientengel wird gemäß den Geräteangaben hergestellt.
Nach vollständiger Polymerisation wird das Gradientengel mit Wasser R gespült. Überschüssige Flüssigkeit wird entfernt. Die Lösung, die die Grundlage für das Anreicherungsgel bildet, wird in die Apparatur eingefüllt und ein sauberer Kamm wird eingelegt. Anschließend lässt man die Lösung des Anreicherungsgels polymerisieren.
Geldimension: 8,6 x 6,7 x 0,1 cm (Breite x Länge x Dicke)
SDS-PAGE-Laufpuffer (10x): 144 g Glycin R, 30 g Tris-Hydrochlorid (= Tris(hydroxymethyl)-aminomethan- bzw. Trometamolhydrochlorid) und 10 g Natriumdodecylsulfat R werden in Wasser R zu 1 000 ml gelöst. Der pH-Wert der Lösung sollte zwischen 8,1 und 8,8 liegen.
SDS-PAGE-Laufpuffer (1x): Vor Gebrauch wird der SDS-PAGE-Laufpuffer (10x) 1 zu 10 mit Wasser R verdünnt.
Tris-Hydrochlorid-Lösung (0,5 mol · l–1): 7,88 g Tris-Hydrochlorid wird mit Wasser R zu 100 ml verdünnt.
Natriumdodecylsulfat-Lösung (10 %): 5 g Natriumdodecylsulfat R werden mit Wasser R zu 50 ml verdünnt.
Bromphenolblau-Lösung (1 %): 0,05 g Bromphenolblau R werden mit Wasser R zu 5 ml verdünnt.
Proben-Pufferlösung: 20 ml Tris-Hydrochlorid-Lösung (0,5 mol · l–1), 16 ml Glycerol 85 % R 1, 32 ml Natriumdodecylsulfat-Lösung (10 %), 8 ml β-Mercaptoethanol R, 4 ml Bromphenolblau-Lösung (1 %) und 40 ml Wasser R werden gemischt.
Stammlösungen: Aus dem Lyophilisat wird eine Lösung hergestellt, die ungefähr 10 mg · ml–1 des Ausgangsmaterials enthält. Es können zusätzliche Verdünnungen wie 1 zu 2 oder 1 zu 5 hergestellt werden.
Untersuchungslösung: 150 µl der entsprechenden Stammlösung werden mit 50 µl Proben-Pufferlösung gemischt (beispielsweise in einem 1,5-ml-Eppendorf-Gefäß). Das Reaktionsgefäß wird verschlossen, in einem Wasserbad 5 min lang bei 95 °C erhitzt und danach auf Raumtemperatur abgekühlt.
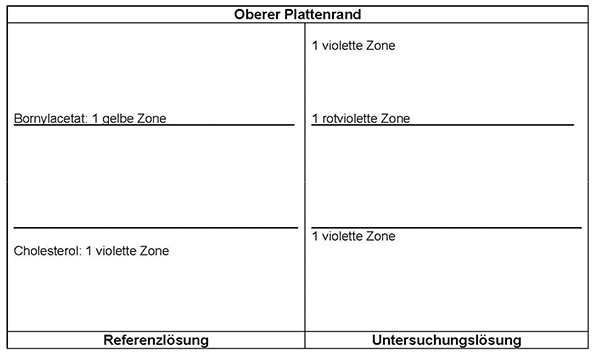
Referenzlösung: Eine Lösung geeigneter Marker für Molekülmassen zur Kalibrierung von SDS-Polyacrylamidgelen im Bereich von 10 bis 250 kDa wird verwendet.
Detektion: Silberfärbung
Fixierlösung: 60 ml wasserfreies Ethanol R werden mit 20 ml wasserfreier Essigsäure R versetzt. Die Mischung wird mit 120 ml Wasser R verdünnt.
Silber-Färbelösung: 2 g Silbernitrat R und 0,2 ml Formaldehyd-Lösung R werden mit Wasser R zu 1 000 ml verdünnt.
Reduktionslösung: 314 mg Natriumthiosulfat R werden mit Wasser R zu 1 000 ml verdünnt.
Entwicklerlösung: 30 g wasserfreies Natriumcarbonat R, 0,5 ml Formaldehyd-Lösung R und 0,1 ml Reduktionslösung werden in Wasser R zu 1 000 ml verdünnt.
Blockierlösung: 5 g Glycin R werden in Wasser R zu 1 000 ml verdünnt.
Die Kassette mit dem Gel wird in die Elektrophorese-Apparatur eingebracht. Danach werden 600 ml des SDS-PAGE-Laufpuffers in die inneren und äußeren Kammern gefüllt. Dabei ist darauf zu achten, dass keine Luftblasen entstehen. Wenn notwendig, muss Puffer nachgefüllt werden. Danach wird der Kamm aus den Geltaschen entfernt und die Taschen mit SDS-PAGE-Laufpuffer gespült, um Luftbläschen oder nicht polymerisierte Acrylamid-Rückstände zu entfernen.
Auftragen: 10 µl Untersuchungslösung und 10 µl Referenzlösung
Laufbedingungen: Die Elektrophorese wird etwa 60 min lang bei Raumtemperatur unter Anlegen von 200 V durchgeführt. Diese Bedingungen gelten bei Verwendung eines Gels im oben genannten Format. Die Elektrophorese wird beendet, sobald die Farbstofffront die Referenzlinie auf der Kassette erreicht.
Der Lauf wird gestartet.
Silberfärbung: Nach dem Lauf wird das Gel aus der Kammer entfernt. Gegebenenfalls wird die Anleitung des Herstellers beachtet.
Folgende Schritte werden ausgeführt, wobei nach den Behandlungen die Lösungen jeweils vorsichtig abdekantiert werden:
- –
-
Fixieren: Das Gel wird 60 min lang unter leichtem Schütteln in Fixierlösung getaucht.
- –
-
Waschen: Das Gel wird 2-mal 20 min lang unter leichtem Schütteln in einer Mischung von 70 ml Wasser R und 30 ml wasserfreiem Ethanol R gewaschen.
- –
-
Waschen: Das Gel wird einmal 20 min lang unter leichtem Schütteln in Wasser R gewaschen.
- –
-
Reduktion: Das Gel wird etwa 1 min lang unter leichtem Schütteln in 100 ml Reduktionslösung getaucht.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Färben: Das Gel wird maximal 20 min lang unter leichtem Schütteln in Silber-Färbelösung getaucht. Das Gel bleibt nur so lange in der Färbelösung, bis sich eindeutige Banden ergeben. Danach kann der Färbeschritt beendet werden, auch wenn 20 min noch nicht abgelaufen sind.
- –
-
Waschen: Das Gel wird 3-mal jeweils etwa 20 s lang unter leichtem Schütteln mit Wasser R gewaschen.
- –
-
Entwicklung: Das Gel wird etwa 3 bis 5 min lang unter leichtem Schütteln in 100 ml Entwicklerlösung getaucht.
- –
-
Waschen: Das Gel wird etwa 20 s lang unter leichtem Schütteln mit 100 ml Wasser R gewaschen.
- –
-
Blockieren: Das Gel wird etwa 10 min lang in Blockierlösung getaucht.
Ergebnis: Der Bereich der Molekülmassen und die Zonenfolge im Elektropherogramm der Untersuchungslösung sind aus den nachstehenden Angaben ersichtlich. Weitere Zonen können vorhanden sein.
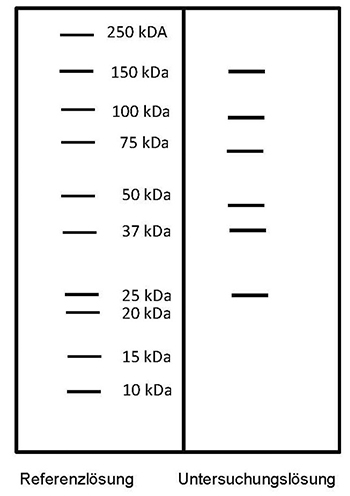
Prüfung auf Reinheit
Polyacrylamid-Gelelektrophorese unter reduzierenden Bedingungen (2.2.31): Die unter „Prüfung auf Identität“ beschriebenen Zonen müssen vorhanden sein. Keine weiteren stark gefärbten Zonen dürfen zu erkennen sein.
Wasser (2.5.12): höchstens 5,0 Prozent, bestimmt mit 50 mg Substanz
Gehaltsbestimmung
Proteingehalt (2.5.33, Methode 4): 100 bis 500 mg · g–1
Untersuchungslösung: 10 mg Lyophilisat werden mit destilliertem Wasser R zu 1 ml verdünnt. Aus dieser Lösung werden weitere Verdünnungen hergestellt (zum Beispiel von 1 mg · ml–1 bis 5 mg · ml–1), sodass die Absorption bei der Messung innerhalb der Kalibrierkurve liegt.
Referenzlösungen: Rinderalbumin R 1 wird mit destilliertem Wasser R zu Konzentrationen im Bereich von 20 μg · ml–1 bis 2 000 μg · ml–1 verdünnt, zum Beispiel 25 μg · ml–1, 125 μg · ml–1, 250 μg · ml–1, 500 μg · ml–1, 750 μg · ml–1, 1 000 μg · ml–1, 1 500 μg · ml–1 und 2 000 μg · ml–1. Es müssen mindestens fünf Referenzlösungen hergestellt werden.
Die Gehaltsbestimmung kann alternativ auch mit einem käuflich erwerbbaren Kit durchgeführt werden.
Lagerung
Dicht verschlossen, bei Raumtemperatur
Arzneiformen
Herstellung
Lösung D1 und flüssige Verdünnungen nach Vorschrift 5a mit Gereinigtem Wasser oder nach Vorschrift 5b mit Wasser für Injektionszwecke
Verreibungen nach Vorschrift 6
Eigenschaften
Die Lösung D1 ist eine gelbbraune Flüssigkeit.
Die 1. Dezimalverreibung ist ein weißes bis hell gelbbraunes Pulver.
Prüfung auf Identität
SDS-Polyacrylamid-Gelelektrophorese (2.2.31): Die Bestimmung erfolgt wie unter „Prüfung auf Identität“ beschrieben mit 100 µl der 1. Dezimalverdünnung oder 100 mg der 1. Dezimalverreibung.
Prüfung auf Reinheit
Trockenrückstand (2.8.16): mindestens 9,0 Prozent für die Lösung D1
Anmerkungen zur Monographie:
- –
-
Definition: Aktualisierung der wissenschaftlichen Pflanzenbezeichnungen
- –
-
Streichung des Geruchs bei Droge und Urtinktur
- –
-
Identitätsprüfung mittels DC: Bei der Untersuchungslösung: Trocknen der organischen Phasen mit wasserfreiem Natriumsulfat R; Aufnahme der HPTLC-Bedingungen und Ersatz der DC-Beschreibung durch eine schematische Darstellung (DC-Kasten)
- –
-
Reinheitsprüfung: Änderung der relativen Dichte aufgrund der vorgelegten Chargendaten
Euphrasia 3c
Euphrasia officinalis 3c
Verwendet werden die frischen ganzen Pflanzen von Euphrasia stricta D. Wolff und Euphrasia officinalis L. und deren Bastarde oder Mischungen davon zur Blütezeit.
Beschreibung
Die Pflanzen haben ein schmächtiges Wurzelwerk mit einer dünnen, verkrümmten Hauptwurzel und wenigen Seitenwurzeln. Die bis 300 mm hohen Stängel steigen aus kurzem Grund straff auf, sie sind fast stielrund, von rückwärts gebogenen, krausen Härchen flaumig und in der Regel auch drüsenhaarig. Sie sind in der Regel braunviolett. Wenn Seitentriebe vorhanden sind, gehen diese ziemlich steil bis rechtwinklig von der Hauptachse ab und steigen bogig auf.
Die 3 bis 17 mm langen Laubblätter sind steif, im Umriss breit keilförmig, rasch in den Blattgrund verschmälert, die oberen mehr eiförmig und spitzer. Die sitzenden Blätter sind unterwärts in der Regel deutlich, oberwärts oft weniger deutlich gegenständig. Sie tragen auf der Unterseite kurze Borsten und in der Regel auch längere, geschlängelte Drüsenhaare (Lupe).
Der Blütenstand ist vielblütig. Die Deckblätter der Blüten sind etwas kürzer und breiter als die obersten Laubblätter und tragen am Rand drei bis sechs spitze oder kurz stachelspitzige Zähne. Die fast ungestielten Blüten haben einen vierzipfeligen, schwach dorsiventralen Kelch. Die insbesondere am Rand drüsenlos oder langdrüsig behaarten Kelchzipfel sind schmal dreieckig und laufen manchmal in eine kurze Stachelspitze aus. Die Krone ist 6 bis 15 mm lang und deutlich zweilippig, außen in der Regel behaart und weiß bis blasslila. Die drei Zipfel der Unterlippe tragen je drei violette Radialstreifen, die Mitte der Unterlippe einen großen, gelben Fleck. Die trichterförmige Kronröhre hat einen gelben Schlund. Die vier Staubblätter besitzen lange, glatte, nach außen gebogene Filamente und dunkle, fest miteinander verklebte Antheren. Der dünne Griffel folgt der Krümmung der Oberlippe und ragt vorn heraus. Er ist im mittleren Teil behaart und trägt eine kleine, kopfige, ockerfarbene Narbe.
Bei Euphrasia stricta und Euphrasia officinalis L. handelt es sich um formenreiche Sammelarten, die in den meisten Merkmalen große Variabilität zeigen.
Arzneiformen
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 3c
Eigenschaften
Die Urtinktur ist eine dunkelbraune Flüssigkeit.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: 10 ml Urtinktur werden mit 10 ml Wasser R versetzt. Die Mischung wird 2-mal mit je 10 ml Ethylacetat R ausgeschüttelt. Die vereinigten organischen Phasen werden mit wasserfreiem Natriumsulfat R getrocknet, durch einen Filter1 filtriert und unter vermindertem Druck zur Trockne eingedampft. Der Rückstand wird in 1 ml Methanol R aufgenommen.
Referenzlösung: 10 mg Rutosid-Trihydrat R, 10 mg Hyperosid R und 10 mg Kaffeesäure R werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R (5 bis 40 μm) [oder DC-Platte mit Kieselgel R (2 bis 10 μm)]
Fließmittel: wasserfreie Ameisensäure R, Wasser R, Ethylacetat R (8:8:84 V/V/V)
Auftragen: 30 µl [oder 8 μl] Untersuchungslösung und 10 µl [oder 2 μl] Referenzlösung; bandförmig 20 mm [oder 8 mm]
Laufstrecke: 15 cm [oder 7 cm]
Trocknen: an der Luft
Detektion: Nach Verdunsten des Fließmittels wird die Platte mit einer Lösung von Diphenylboryloxyethylamin R (10 g · l−1) in Methanol R und anschließend mit einer Lösung von Macrogol 400 R (50 g · l−1) in Methanol R behandelt. Die Auswertung erfolgt nach 30 min im ultravioletten Licht bei 365 nm.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere fluoreszierende Zonen vorhanden sein.
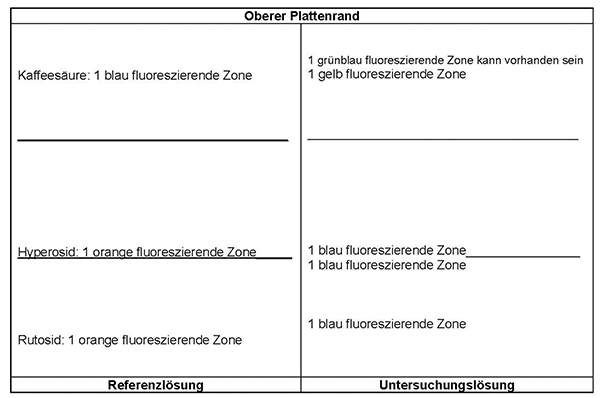
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,960 bis 0,980
Trockenrückstand (H 2.2.6): mindestens 2,0 Prozent
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Unter Prüfung auf Reinheit ist für die Bestimmung der Ginkgolsäuren die Berechnungsformel aufgenommen worden. Der bisherige Verweis im HAB auf die Formel in der Ph.-Eur.-Monographie „Quantifizierter, raffinierter Ginkgotrockenextrakt“ war nicht korrekt, da für die Bestimmung in der Urtinktur eine höher verdünnte Untersuchungslösung eingesetzt wird als für den Ginkgotrockenextrakt.
Ginkgo biloba
Ginkgo
Verwendet werden die frischen Blätter von Ginkgo biloba L.
Beschreibung
Die Blätter haben einen 20 bis 90 mm langen, oberseits abgeplatteten oder schwach rinnigen Blattstiel, der sich erst allmählich, dann rasch zu einer fächerförmigen, in der Regel zweilappigen, sonst schwach mehrlappigen Blattspreite erweitert. Diese ist 50 bis 80 mm lang, 60 bis 100 mm breit, derb ledrig, beiderseits kahl, oberseits mittel- bis dunkelgrün, unterseits wenig heller. Die stumpf- oder breit-kegelförmige Spreite hat keinen Mittelnerv, sondern ist bis zum Blattrand von regelmäßig dichotom verzweigten Adern durchzogen. Der Blattrand ist apikal unregelmäßig gebuchtet oder mehr oder weniger tief eingeschnitten und an den Seiten ganzrandig.
Arzneiformen
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 3a
Eigenschaften
Die Urtinktur ist eine grünbraune Flüssigkeit.
Prüfung auf Identität
Dünnschichtchromatographie (2.2.27)
Untersuchungslösung: die Urtinktur
Referenzlösung: 10 mg Rutosid-Trihydrat R, 5 mg Hyperosid R und 5 mg Quercetin-Dihydrat R werden in 10 ml Methanol R gelöst.
Platte: DC-Platte mit Kieselgel R
Fließmittel: wasserfreie Ameisensäure R, Wasser R, Ethylacetat R (10:10:80 V/V/V)
Auftragen: 20 µl Untersuchungslösung und 10 µl Referenzlösung; bandförmig 20 mm
Laufstrecke: 15 cm
Detektion: Nach Verdunsten des Fließmittels wird die Platte mit einer Lösung von Diphenylboryloxyethylamin R (10 g · l−1) in Methanol R und anschließend mit einer Lösung von Macrogol 400 R (50 g · l−1) in Methanol R behandelt. Die Auswertung erfolgt nach 30 min im ultravioletten Licht bei 365 nm.
Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere fluoreszierende Zonen vorhanden sein.

Prüfung auf Reinheit
Ginkgolsäuren (2.2.29): höchstens 2500 ppm
Untersuchungslösung: 1,00 g Urtinktur wird mit 100,0 ml Methanol R verdünnt.
Die weitere Bestimmung erfolgt wie in der Monographie Quantifizierter, raffinierter Ginkgotrockenextrakt (Ph. Eur.) unter „Gehaltsbestimmung, Ginkgolsäuren“ angegeben.
Der Gehalt in ppm an Ginkgolsäuren wird als Gehalt in ppm an Ginkgolsäure C17 nach folgender Formel berechnet:
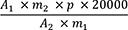
- A1
-
= Summe der Flächen der Peaks der Ginkgolsäuren C13, C15 und C17 im Chromatogramm der Untersuchungslösung
- A2
-
= Fläche des Peaks der Ginkgolsäure C17 im Chromatogramm der Referenzlösung
- m1
-
= Einwaage der Urtinktur zur Herstellung der Untersuchungslösung in Gramm
- m2
-
= Masse von Ginkgolsäuren CRS zur Herstellung der Referenzlösung in Gramm
- p
-
= Prozentgehalt an Ginkgolsäure C17 in Ginkgolsäuren CRS
Relative Dichte (2.2.5): 0,905 bis 0,925
Trockenrückstand (H 2.2.6): mindestens 3,5 Prozent
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Aufgrund der beabsichtigten Streichung des Reagenzes „Karl-Fischer-Lösung R“ in der
Ph. Eur. wird bei der Reinheitsprüfung auf Wasser (2.5.12, Methode B) „Karl-Fischer-Lösung R“ durch „kommerzielle Fertiglösung für die Karl-Fischer-Bestimmung“ ersetzt. Außerdem wird die Rührzeit von 2 Stunden auf 15 Minuten verkürzt, da diese Rührdauer für die Prüfung ausreichend ist.
Hydrargyrum nitricum oxydulatum
Mercurius nitricus oxydulatus
Hg2(NO3)2 · 2 H2O
M r 561,2
Verwendet wird Quecksilber(I)-nitrat, das mindestens 94,0 und höchstens 100,5 Prozent Hg2(NO3)2 · 2 H2O enthält.
Eigenschaften
Farblose, lichtempfindliche hygroskopische Kristalle; die Substanz zersetzt sich in Wasser unter Bildung eines unlöslichen basischen Salzes; leicht löslich in verdünnter Salpetersäure.
Die Substanz schmilzt bei etwa 66 °C.
Prüfung auf Identität
- A.
-
Wird die Substanz mit verdünnter Natriumhydroxid-Lösung R versetzt, färbt sie sich schwarz.
- B.
-
0,1 g Substanz werden in einer Mischung von 3 ml Wasser R und 0,1 ml Salpetersäure R gelöst. Wird die Lösung mit Salzsäure R versetzt, entsteht ein weißer Niederschlag, der sich nach Zusatz von Ammoniak-Lösung R schwarz färbt.
- C.
-
50 mg Substanz werden mit 5 ml Essigsäure R versetzt. Die Mischung wird kurz erwärmt und nach dem Abkühlen filtriert. Wird das Filtrat vorsichtig mit 0,5 ml Diphenylamin-Lösung R unterschichtet, färbt sich die Berührungsfläche der Schichten blau.
Prüfung auf Reinheit
Prüflösung: 5,0 g Substanz werden nach Zusatz von 0,5 ml Salpetersäure R in 25 ml destilliertem Wasser R gelöst. Die Lösung wird mit 4 ml wasserfreier Ameisensäure R und 12 ml konzentrierter Ammoniak-Lösung R versetzt und die Mischung 30 min lang auf dem Wasserbad erwärmt. Nach dem Abkühlen wird die Mischung filtriert und das Filtrat unter Nachwaschen des Filters mit destilliertem Wasser R zu 50,0 ml verdünnt.
Aussehen der Lösung: Die Prüflösung muss klar (2.2.1) und farblos (2.2.2, Methode II) sein.
Chlorid (2.4.4): 10,0 ml Prüflösung werden mit Wasser R zu 15,0 ml verdünnt. Die Lösung muss der Grenzprüfung auf Chlorid entsprechen (50 ppm).
Sulfat (2.4.13): 10,0 ml Prüflösung werden mit destilliertem Wasser R zu 15,0 ml verdünnt. Die Lösung muss der Grenzprüfung auf Sulfat entsprechen (150 ppm).
Quecksilber(II)-Verbindungen: höchstens 2,5 Prozent, berechnet als Hg2(NO3)2 · 2 H2O
0,500 g Substanz werden in einer Mischung von 0,5 ml Salpetersäure R und 50 ml Wasser R gelöst und die Lösung mit 0,5 ml Salzsäure R versetzt. Nach 15 min wird die Mischung filtriert. Das Filter wird 3-mal mit je 10 ml Wasser R gewaschen. Das mit der Waschflüssigkeit vereinigte Filtrat wird mit verdünnter Natriumhydroxid-Lösung R unter Verwendung von 0,05 ml Methylorange-Lösung R als Indikator neutralisiert. Nach Zusatz von 10,0 ml Natriumedetat-Lösung (0,01 mol · l−1) wird die Mischung 5 min lang stehen gelassen, anschließend mit 5 ml Pufferlösung pH 10,9 R und 50 mg Eriochromschwarz-T-Mischindikator RN versetzt und mit Zinksulfat-Lösung (0,01 mol · l−1) bis zum Farbumschlag nach Rot titriert.
1 ml Natriumedetat-Lösung (0,01 mol · l−1) entspricht 2,806 mg Hg2(NO3)2 · 2 H2O.
Wasser (2.5.12, Methode B): mindestens 5,0 und höchstens 9,0 Prozent, mit 0,500 g Substanz nach der Karl-Fischer-Methode bestimmt
Nach Zusatz einer kommerziellen Fertiglösung für die Karl-Fischer-Bestimmung wird die Lösung 15 min lang gerührt.
Gehaltsbestimmung
0,150 g Substanz werden mit 2 ml Salpetersäure R versetzt. Die Mischung wird zum Sieden erhitzt, bis eine Lösung entstanden ist, und die Lösung nach dem Abkühlen mit 50 ml Wasser R verdünnt. Die Lösung wird mit verdünnter Natriumhydroxid-Lösung R unter Verwendung von 0,05 ml Methylorange-Lösung R als Indikator neutralisiert, mit 10,0 ml Natriumedetat-Lösung (0,1 mol · l−1) versetzt und die Mischung 5 min lang stehen gelassen. Nach Zusatz von 5 ml Pufferlösung pH 10,9 R und 50 mg Eriochromschwarz-T-Mischindikator RN wird mit Zinksulfat-Lösung (0,1 mol · l−1) bis zum Farbumschlag nach Rot titriert.
1 ml Natriumedetat-Lösung (0,1 mol · l−1) entspricht 28,06 mg Hg2(NO3)2 · 2 H2O.
Der aus dem Verbrauch an Natriumedetat-Lösung (0,1 mol · l−1) berechnete Gehalt muss um den unter „Prüfung auf Reinheit“ ermittelten Gehalt an Quecksilber(II)-Verbindungen, berechnet als Hg2(NO3)2 · 2 H2O, vermindert werden.
Arzneiformen
Die 1. Dezimalverreibung enthält mindestens 8,9 und höchstens 10,6 Prozent Hg2(NO3)2 · 2 H2O.
Herstellung
Verreibungen nach Vorschrift 6
Eigenschaften
Die 1. Dezimalverreibung ist ein weißes Pulver.
Prüfung auf Identität
- A.
-
1 g der 1. Dezimalverreibung wird in einer Mischung von 5 ml Wasser R und 0,1 ml Salpetersäure R gelöst. Die Lösung gibt die Identitätsreaktion B der Substanz.
- B.
-
0,5 g der 1. Dezimalverreibung geben die Identitätsreaktion C der Substanz.
Gehaltsbestimmung
1,50 g der 1. Dezimalverreibung werden in 10 ml einer Lösung, die 5 g Natriumchlorid R und 5 mg Natriumdodecylsulfat R in 100 ml enthält, suspendiert und die Mischung wird zentrifugiert. Die überstehende Lösung wird verworfen und der Waschvorgang mit oben beschriebener Lösung 3-mal wiederholt. Der Rückstand wird in 2 ml Salpetersäure R unter Erwärmen gelöst. Nach dem Abkühlen wird die Lösung mit 2 ml Wasser R verdünnt und die Mischung unter Nachspülen mit Wasser R quantitativ in einen Erlenmeyerkolben, der 50 ml Wasser R enthält, überführt. Die Mischung wird mit verdünnter Natriumhydroxid-Lösung R unter Verwendung von 0,05 ml Methylorange-Lösung R als Indikator neutralisiert, mit 10,0 ml Natriumedetat-Lösung (0,1 mol · l−1) versetzt und 5 min lang stehen gelassen. Nach Zusatz von 5 ml Pufferlösung pH 10,9 R und 50 mg Eriochromschwarz-T-Mischindikator RN wird mit Zinksulfat-Lösung (0,1 mol · l−1) bis zum Farbumschlag nach Rot titriert.
1 ml Natriumedetat-Lösung (0,1 mol · l−1) entspricht 28,06 mg Hg2(NO3)2 · 2 H2O.
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Bei der Gehaltsbestimmung wurde bei der Referenzlösung und der mobilen Phase die chloridhaltige Pufferlösung pH 2,0 R durch eine 0,2 molare Phosphat-Pufferlösung pH 2,0 ersetzt, um die Messempfindlichkeit der Bestimmung zu verbessern. Die Pufferlösung wird als Reagenz in das HAB aufgenommen.
Aufgrund der o. g. Änderung wurden die Retentionszeit für α-Solanin und die relativen Retentionen für die Steroidalkaloide der Untersuchungslösung angepasst und ein neues Beispielchromatogramm für die Untersuchungslösung aufgenommen.
Solanum dulcamara
Dulcamara
Verwendet werden die frischen Triebe von Solanum dulcamara L. vor der Blütezeit.
Beschreibung
Die Triebe haben, insbesondere beim Zerreiben, einen unangenehmen Geruch. Der junge, häufig kletternde Stängel der halbstrauchartigen Pflanze ist krautig, rundlich bis etwas kantig, bis etwa 5 mm dick, innen hohl, außen grün und in der Regel kahl, bisweilen verstreut oder mehr oder weniger abstehend bis angedrückt filzig behaart. Die wechselständigen, in der Regel 50 bis 90 mm, selten 30 mm langen und in der Regel 25 bis 50 mm, selten 15 mm breiten, bis 30 mm lang gestielten Laubblätter sind sehr unterschiedlich ausgebildet. Ihre Spreite ist gewöhnlich eiförmig-lanzettlich, spitz oder zugespitzt, am Grund herz-, bisweilen auch spießförmig, ganzrandig oder etwas wellig, bisweilen am Grund mit einem oder zwei, selten bis vier Lappen oder kleinen, gestielten Fiederblättchen versehen. Ihre Fläche ist beiderseits blassgrün bis bläulich grün, kahl oder insbesondere unterseits auf den Nerven verstreut oder filzig behaart.
Arzneiformen
Die Urtinktur enthält mindestens 0,050 und höchstens 0,250 Prozent Steroidalkaloide, berechnet als α-Solanin (C45H73NO15; Mr 868,1).
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 2a
Eigenschaften
Die Urtinktur ist eine gelbbraune bis grünbraune Flüssigkeit.
Prüfung auf Identität
- A.
-
Dünnschichtchromatographie (2.2.27)Untersuchungslösung: die UrtinkturReferenzlösung: 10 mg α-Solanin RH und 10 mg Papaverinhydrochlorid R werden in 10 ml Methanol R gelöst.Platte: DC-Platte mit Kieselgel RFließmittel: Essigsäure 99 % R, 1-Butanol R, Wasser R (10:40:50 V/V/V); die obere Phase wird verwendet.Auftragen: 40 µl Untersuchungslösung und 10 µl Referenzlösung; bandförmig 20 mmLaufstrecke: 10 cmTrocknen: im WarmluftstromDetektion: Nach Verdunsten des Fließmittels wird die Platte mit Dragendorffs Reagenz R und anschließend mit Natriumnitrit-Lösung R behandelt. Die Auswertung erfolgt im Tageslicht.Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.

- B.
-
Die unter „Gehaltsbestimmung“ erhaltenen Chromatogramme werden ausgewertet. Die im HPLC-Beispielchromatogramm aufgeführten vier Alkaloid-Peaks treten bei den relativen Retentionszeiten von etwa 0,7 bis 0,8; etwa 0,8 bis 0,9; etwa 0,8 bis 1,0 und etwa 1,0 bis 1,2, bezogen auf α-Solanin, auf.
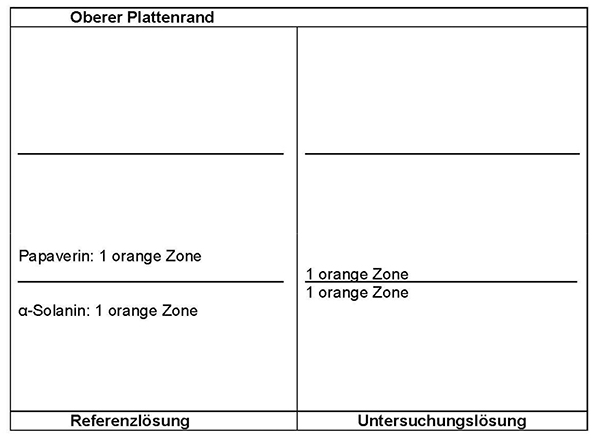
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,935 bis 0,955
Trockenrückstand (H 2.2.6): mindestens 2,0 Prozent
Gehaltsbestimmung
Flüssigchromatographie (2.2.29)
Untersuchungslösung: 4,0 g Urtinktur werden in einen 5,0-ml-Messkolben eingewogen, mit 0,1 ml Salzsäure R versetzt und mit Ethanol 96 % R zu 5,0 ml aufgefüllt.
Probenaufbereitung: Eine Kartusche, gefüllt mit Kationenaustauscher RH (60 µm; 150 mg) wird mit 1,0 ml Methanol R und 1,0 ml Wasser R konditioniert. Anschließend werden 0,250 ml Untersuchungslösung auf die Säule gegeben. Die Säule wird nacheinander mit je 1,0 ml Salzsäure (0,1 mol · l−1), Wasser R und Methanol R gewaschen. Anschließend wird sie 2-mal mit je 1,0 ml einer Mischung von 5 Volumteilen konzentrierter Ammoniak-Lösung R und 95 Volumteilen Methanol R eluiert. Das Eluat wird in einem 2,0-ml-Messkolben aufgefangen und mit einer Mischung von 5 Volumteilen konzentrierter Ammoniak-Lösung R und 95 Volumteilen Methanol R zu 2,0 ml aufgefüllt.
Referenzlösung: 5,0 mg α-Solanin RH werden in einer Mischung gleicher Volumteile Methanol R und Phosphat-Pufferlösung pH 2,0 (0,2 mol · l–1) RH zu 10,0 ml gelöst.
2,0 ml Lösung werden mit einer Mischung gleicher Volumteile Methanol R und Phosphat-Pufferlösung pH 2,0 (0,2 mol · l–1) RH zu 10,0 ml verdünnt.
Säule
- –
-
Größe: l = 0,100 m, ⌀ = 4,6 mm
- –
-
Stationäre Phase: nachsilanisiertes, octadecylsilyliertes Kieselgel zur Chromatographie mit festem Kern R (2,6 µm)2
- –
-
Temperatur: 23 °C
Mobile Phase: Phosphat-Pufferlösung pH 2,0 (0,2 mol · l–1) RH, Acetonitril zur Chromatographie R (79:21 V/V)
Durchflussrate: 1,5 ml · min−1
Detektion: Spektrometer bei 203 nm
Einspritzen: 20 µl
Relative Retention (bezogen auf α-Solanin, tR etwa 8 bis 9 min)
- Steroidalkaloid I:
-
etwa 0,7 bis 0,8
- Steroidalkaloid II:
-
etwa 0,8 bis 0,9
- Steroidalkaloid III:
-
etwa 0,8 bis 1,0
- Steroidalkaloid IV:
-
etwa 1,0 bis 1,2
Chromatographiedauer: 20 min
Eignungsprüfung
- –
-
Wiederholpräzision: höchstens 2,0 Prozent relative Standardabweichung für die Flächen des α-Solanin-Peaks nach 6 Einspritzungen der Referenzlösung
Der Prozentgehalt (m/m) an Steroidalkaloiden wird nach folgender Formel berechnet:
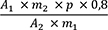
- A1
-
= Summe der Peakflächen der Steroidalkaloide im Chromatogramm der Untersuchungslösung
- A2:
-
Fläche des α-Solanin-Peaks im Chromatogramm der Referenzlösung
- m1:
-
Einwaage der Urtinktur in Gramm
- m2:
-
Masse der Referenzsubstanz α-Solanin RH in Gramm
- p
-
= Prozentgehalt an α-Solanin in α-Solanin RH
Beispielchromatogramm: Untersuchungslösung Solanum dulcamara ⌀
Lagerung
Vor Licht geschützt
Anmerkungen zur Monographie:
Bei der Gehaltsbestimmung wurde bei der Referenzlösung und der mobilen Phase die chloridhaltige Pufferlösung pH 2,0 R durch eine 0,2 molare Phosphat-Pufferlösung pH 2,0 ersetzt, um die Messempfindlichkeit der Bestimmung zu verbessern. Die Pufferlösung wird als Reagenz in das HAB aufgenommen.
Aufgrund der o. g. Änderung wurden die Retentionszeit für α-Solanin und die relativen Retentionen für die Steroidalkaloide der Untersuchungslösung angepasst und ein neues Beispielchromatogramm für die Untersuchungslösung aufgenommen.
Solanum nigrum
Verwendet werden die frischen ganzen Pflanzen von Solanum nigrum L. zur Blütezeit.
Beschreibung
Die Pflanzen entwickeln beim Zerreiben einen schwachen, dumpfen Geruch.
Von der elfenbeinfarbigen, einfachen oder verzweigten, bis 200 mm langen und bis höchstens 10 mm dicken Hauptwurzel gehen zahlreiche, unterschiedlich dicke und lange Seitenwurzeln ab. Der aufrechte Stängel ist bis 0,5 m, gelegentlich bis 0,7 m hoch, grün bis dunkelgrün, verzweigt, im unteren Teil rund und im oberen mehr oder weniger kantig. Er ist kahl oder mit verstreut stehenden, kurzen, anliegenden bis etwas abstehenden, einwärts gekrümmten, bis etwa 0,5 mm langen Haaren besetzt.
Die dicklichen Blätter sind gestielt, breit-eiförmig, elliptisch-eiförmig oder fast dreieckig, bis 70 mm, selten bis 130 mm lang und bis 60 mm, selten bis 85 mm breit, am oberen Ende zugespitzt, seltener spitz und mit keilförmigem oder gestutztem, etwas in den Stiel verschmälertem Blattgrund. Die Blätter sind kahl oder oberseits mit verstreut vorkommenden, unterseits mit insbesondere auf den Nerven stehenden hakenförmigen Borstenhaaren besetzt. Der Blattrand ist glatt oder unterhalb der Blattspitze dreimal, seltener vier- bis fünfmal breit buchtig gezähnt, selten stärker gezähnt.
Die Blüten stehen zu vier bis sechs in doldenartigen Wickeln auf 8 bis 20 mm langen, rückwärts gebogenen Blütenstielen, die kahl oder mit rückwärts gekrümmten Haaren besetzt sind. Der bleibende, kahle oder verstreut behaarte Kelch hat fünf kurze, stumpf-eiförmige Zipfel. Die mehr oder weniger radförmig ausgebreitete, weiße, verwachsene Blütenkrone ist mit einem Durchmesser von 6 bis 7 mm zwei- bis dreimal länger als der Kelch und bis zu zwei Drittel in fünf stumpf-eiförmige oder eiförmig-lanzettliche, außen fein behaarte Zipfel geteilt. Die fünf zusammenneigenden, goldgelben, auf kurzen Staubfäden stehenden Staubblätter bilden einen die Krone überragenden Kegel. Der Fruchtknoten ist rund bis eiförmig, zwei- bis dreimal länger als die Staubblätter und wird von einem in den unteren zwei Dritteln fein behaarten Griffel gekrönt. Die in einigen Blütenständen schon während der Blütezeit erkennbaren grünen Beeren entwickeln sich zu 8 bis 10 mm großen, schwarz glänzenden Früchten.
Arzneiformen
Die Urtinktur enthält mindestens 0,005 und höchstens 0,050 Prozent Steroidalkaloide, berechnet als α-Solanin (C45H73NO15; Mr 868,1).
Herstellung
Urtinktur und flüssige Verdünnungen nach Vorschrift 2a
Eigenschaften
Die Urtinktur ist eine bräunlich gelbe Flüssigkeit.
Prüfung auf Identität
- A.
-
Dünnschichtchromatographie (2.2.27)Untersuchungslösung: die UrtinkturReferenzlösung: 10 mg α-Solanin RH und 10 mg Papaverinhydrochlorid R werden in 10 ml Methanol R gelöst.Platte: DC-Platte mit Kieselgel RFließmittel: Essigsäure 99 % R, 1-Butanol R, Wasser R (10:40:50 V/V/V); die obere Phase wird verwendet.Auftragen: 40 µl Untersuchungslösung und 10 µl Referenzlösung; bandförmig 20 mmLaufstrecke: 10 cmTrocknen: im WarmluftstromDetektion: Nach Verdunsten des Fließmittels wird die Platte mit Dragendorffs Reagenz R und anschließend mit Natriumnitrit-Lösung R behandelt. Die Auswertung erfolgt im Tageslicht.Ergebnis: Die Zonenfolge in den Chromatogrammen von Referenzlösung und Untersuchungslösung ist aus den nachstehenden Angaben ersichtlich. Im Chromatogramm der Untersuchungslösung können weitere Zonen vorhanden sein.
- B.
-
Die unter „Gehaltsbestimmung“ erhaltenen Chromatogramme werden ausgewertet. Die im HPLC-Beispielchromatogramm aufgeführten zwei Alkaloid-Peaks treten bei den relativen Retentionszeiten von etwa 0,8 bis 0,9 und etwa 1,0 bis 1,2, bezogen auf α-Solanin, auf.
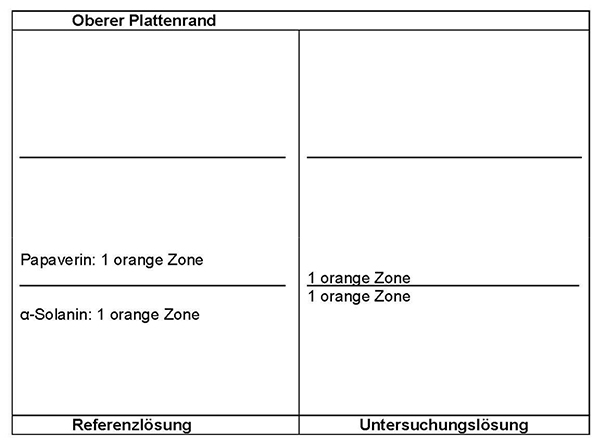
Prüfung auf Reinheit
Relative Dichte (2.2.5): 0,930 bis 0,950
Trockenrückstand (H 2.2.6): mindestens 1,2 Prozent
Gehaltsbestimmung
Flüssigchromatographie (2.2.29)
Untersuchungslösung: 4,0 g Urtinktur werden in einen 5,0-ml-Messkolben eingewogen, mit 0,1 ml Salzsäure R versetzt und mit Ethanol 96 % R zu 5,0 ml aufgefüllt.
Probenaufbereitung: Eine Kartusche, gefüllt mit Kationenaustauscher RH (60 µm; 150 mg) wird mit 1,0 ml Methanol R und 1,0 ml Wasser R konditioniert. Anschließend werden 0,250 ml Untersuchungslösung auf die Säule gegeben.
Die Säule wird nacheinander mit je 1,0 ml Salzsäure (0,1 mol · l−1), Wasser R und Methanol R gewaschen. Anschließend wird sie 2-mal mit je 1,0 ml einer Mischung von 5 Volumteilen konzentrierter Ammoniak-Lösung R und 95 Volumteilen Methanol R eluiert. Das Eluat wird in einem 2,0-ml-Messkolben aufgefangen und mit einer Mischung von 5 Volumteilen konzentrierter Ammoniak-Lösung R und 95 Volumteilen Methanol R zu 2,0 ml aufgefüllt.
Referenzlösung: 5,0 mg α-Solanin RH werden in einer Mischung gleicher Volumteile Methanol R und Phosphat-Pufferlösung pH 2,0 (0,2 mol · l–1) RH zu 10,0 ml gelöst. 2,0 ml Lösung werden mit einer Mischung gleicher Volumteile Methanol R und Phosphat-Pufferlösung pH 2,0 (0,2 mol · l–1) RH zu 10,0 ml verdünnt.
Säule
- –
-
Größe: l = 0,100 m, ⌀ = 4,6 mm
- –
-
Stationäre Phase: nachsilanisiertes, octadecylsilyliertes Kieselgel zur Chromatographie mit festem Kern R (2,6 µm)3
- –
-
Temperatur: 23 °C
Mobile Phase: Phosphat-Pufferlösung pH 2,0 (0,2 mol · l–1) RH, Acetonitril zur Chromatographie R (79:21 V/V)
Durchflussrate: 1,5 ml · min−1
Detektion: Spektrometer bei 203 nm
Einspritzen: 20 µl
Relative Retention (bezogen auf α-Solanin, tR etwa 8 bis 9 min)
- Steroidalkaloid I:
-
etwa 0,8 bis 0,9
- Steroidalkaloid II:
-
etwa 1,0 bis 1,2
Chromatographiedauer: 20 min
Eignungsprüfung
- –
-
Wiederholpräzision: höchstens 2,0 Prozent relative Standardabweichung für die Flächen des α-Solanin-Peaks nach 6 Einspritzungen der Referenzlösung
Der Prozentgehalt (m/m) an Steroidalkaloiden wird nach folgender Formel berechnet:
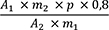
- A1
-
= Summe der Peakflächen der Steroidalkaloide im Chromatogramm der Untersuchungslösung
- A2:
-
Fläche des α-Solanin-Peaks im Chromatogramm der Referenzlösung
- m1:
-
Einwaage der Urtinktur in Gramm
- m2:
-
Masse der Referenzsubstanz α-Solanin RH in Gramm
- p
-
= Prozentgehalt an α-Solanin in α-Solanin RH
Beispielchromatogramm: Untersuchungslösung Solanum nigrum ⌀
Lagerung
Vor Licht geschützt
Neues Reagenz
Phosphat-Pufferlösung pH 2,0 (0,2 mol · l−1) RH
27,22 g Kaliumdihydrogenphosphat R werden in 1 000 ml Wasser R gelöst.
Der pH-Wert (2.2.3) wird mit Phosphorsäure 85 % R eingestellt.
- 1
- geeignet ist z. B. ein Faltenfilter von Macherey-Nagel 614 ¼ (Dicke 0,25 mm, ⌀ 110 mm)
- 2
- geeignet ist z. B. eine Kinetex C18 der Fa. Phenomenex
- 3
- geeignet ist z. B. eine Kinetex C18 der Fa. Phenomenex








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kommentar hinterlassen